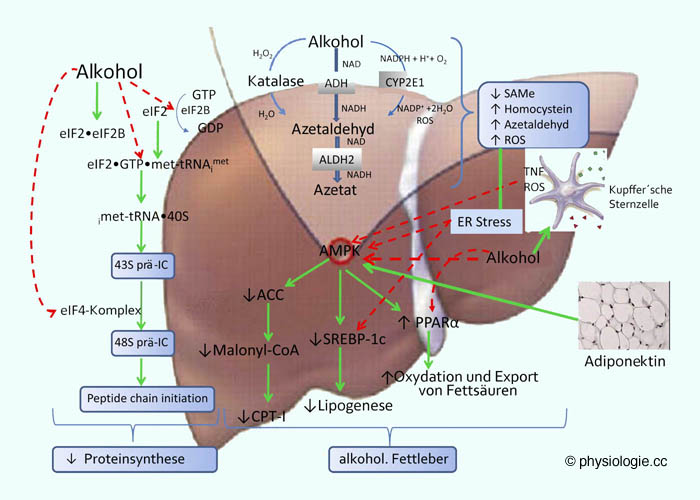
 Abbildung: Hepatischer Abbau von Äthylalkohol und Auswirkungen chronischen Alkoholkonsums
Abbildung: Hepatischer Abbau von Äthylalkohol und Auswirkungen chronischen Alkoholkonsums
Nach
Molina PE, Alcohol - intoxicating roadblocks and bottlenecks in hepatic
protein and lipid metabolism. Amer J Physiol - Endocrin Metab 2008;
295: E1-2
Grüne Pfeile: Anregung, rote unterbrochene Pfeile: Inhibition
Abkürzungen und Beschreibung s. Text

Die Leber oxidiert Alkohol zu Azetaldehyd (Abbildung).
Das erfolgt auf mehreren Wegen, und zwar - bedeutungsmäßig in dieser
Reihenfolge - über die Alkohol-Dehydrogenase (ADH - zytosomal), Zytochrom P-450 (CYP2E1 - endoplasmatisches Retikulum), und Katalase (Peroxisomen).
 Die Alkohol-Dehydrogenase (ADH
- Klasse I-Isoform) ist ein zytosomales Enzym, das sich in der Leber und auch
in der Magenschleimhaut findet (Abbau schon vor Eintritt in die
Blutbahn, begünstigt durch längere Verweildauer) und unterschiedlich
ausgeprägt wird (z.B.im Schnitt effektivere Enzymaktivität bei Männern
im Vergleich zu Frauen, bei europäischstämmigen Menschen im Vergleich
zu asiatischstämmigen).
Die Alkohol-Dehydrogenase (ADH
- Klasse I-Isoform) ist ein zytosomales Enzym, das sich in der Leber und auch
in der Magenschleimhaut findet (Abbau schon vor Eintritt in die
Blutbahn, begünstigt durch längere Verweildauer) und unterschiedlich
ausgeprägt wird (z.B.im Schnitt effektivere Enzymaktivität bei Männern
im Vergleich zu Frauen, bei europäischstämmigen Menschen im Vergleich
zu asiatischstämmigen).
Das mikrosomale ethanoloxidierende System
(MEOS) kann den Abbau weiter steigern. MEOS ist durch mehrmalige
Induktion "trainierbar", der Abbau von Alkohol erfolgt beschleunigt.
Die Metabolisierung von Äthanol liefert 29 kJ/g (7 Cal/g)
Energie (zum Vergleich: Kohlenhydrate und Proteine 17 kJ/g bzw. 4,1
Cal/g, Fette 37 kJ/g bzw. 9,3 Cal/g). Seine Energiedichte ist damit
relativ hoch - sie liegt zwischen der von Fetten einerseits und
Kohlenhydraten andererseits.
Azetaldehyd wird dann durch mitochondriale Aldehyd-Dehydrogenase (ALDH2)
zu Azetat abgebaut. Produkte dieses metabolischen Weges senken den
zellulären Gehalt am Methioninprodukt und Methyldonor
S-Adenosylmethionin (SAMe),
das Methyltransferasen als Substrat brauchen. Gleichzeitig steigt der
Spiegel an Homocystein, Azetaldehyd und Sauerstoffradikalen (ROS). Das bedingt eine Stressreaktion im endoplasmatischen Retikulum (ER stress): Es kommt zu Aktivierung von Promotor-Sequenzen, die als sterol regulatory element-binding proteins (SREBP-1c und -2c) bezeichnet werden, dies führt zu Anhäufung von Triglyzeriden.
Die Aktivität des metabolischen Schlüsselenzyms AMP kinase (AMPK) steht unter mehrfachem alkoholbedingten Einfluss:
 AMPK wird durch Alkohol, endoplasmatischen Stress, Tumornekrosefaktor (TNF) und ROS gehemmt;
AMPK wird durch Alkohol, endoplasmatischen Stress, Tumornekrosefaktor (TNF) und ROS gehemmt;
Adiponektin aus dem Fettgewebe fördert hingegen seine Tätigkeit (chronischer Alkoholkonsum hemmt die Freisetzung von Adiponektin).
AMPK fördert Fettsäureoxydation und -transport durch Aktivierung des nukleären Hormonrezeptors peroxisome proliferator-activated receptor-α (PPARα), unterdrückt SREBP-1c (was die Lipogenese hemmt), und hemmt die Azetyl-CoA-Karboxylase (ACC).
Letzteres senkt die Synthese und steigert die Oxydation von Fettsäuren
(via Absinken des Malonyl-CoA-Spiegels und der Aktivität von Carnitin-Palmitoyltransferase I (CPT I).
Insgesamt führen alkoholbedingte Veränderungen zu beeinträchtigtem Fettstoffwechsel und Fettleber. Auch die hepatische Proteinsynthese (peptide chain initiation) ist betroffen - z.B. der Wechsel zwischen aktiven und inaktiven Formen des eIF2·eIF2B-Komplexes, was die Bildung des
43S-Präinitiationskomplexes (43S prä-IC)
verhindert (dieser hilft beim Rekrutieren ribosomaler 40S-Einheiten an
die mRNS). Chronischer Alkoholkonsum erweitert diesen Defekt auf die
Fähigkeit des elF4-Komplexes, die Anbindung zwischen 43S-Komplex und
5'-Ende der mRNS zu ermöglichen - was die Eiweißsynthese weiter beeinträchtigt.
 Zur Steuerung der Leberfunktion s. dort
Zur Steuerung der Leberfunktion s. dort


 © Helmut
Hinghofer-Szalkay
© Helmut
Hinghofer-Szalkay