Elektro-mechanische Koppelung und Herzschlag  Lusitropie Mechanismus und Steuerung der Kontraktion Ruhedehnungskurve, Druck-Volumen-Beziehung Belastung und Kontraktionsgeschwindigkeit
Lusitropie Mechanismus und Steuerung der Kontraktion Ruhedehnungskurve, Druck-Volumen-Beziehung Belastung und Kontraktionsgeschwindigkeit

Diaden  Calciuminduzierte Calciumfreisetzung (CICR)
Calciuminduzierte Calciumfreisetzung (CICR)
 Praktische Aspekte
Praktische Aspekte  Core messages
Core messages
Aktionspotentiale,
die über das myokardiale Synzytium laufen, lösen Kontraktionen des
Herzmuskels (Systolen) aus. Eine Schlüsselrolle spielen Calciumionen,
die teils aus dem Extrazellulärraum, teils aus intrazellulären
Speichern blitzartig in das Sarkoplasma der Herzmuskelzellen einströmen
und dort die Interaktion kontraktiler Filamente aktivieren. Die
resultierende Verknüpfung von elektrischen (Erregung) und mechanischen
Effekten (Kontraktion) nennt man elektromechanische Kopplung (excitation-contraction coupling).
Die elektromechanische Koppelung verknüpft Aktionspotential und Kontraktion
 Herzmuskelzellen kontrahieren, wenn in ihrem Zytosol der Ca++-Spiegel auf >100 nM ansteigt. Zu einem Viertel beruht dieser Anstieg auf dem Einstrom von Ca++ durch die erregte Zellmembran, zu ~75% stammt er aus dem intrazellulären Speicher: Das Aktionspotential öffnet am Ende von transversalen (T-) Tubuli Ca++-Kanäle in der Nähe des sarkoplasmatischen Retikulums. An dessen Membran regt der Ca++-Einstrom aus den T-Tubuli Calciumkanäle (Ryanodinrezeptoren, RyR) in der Membran des
sarkoplasmatische Retikulums an. Diese Koppelung nennt man
calciuminduzierte Calciumfreisetzung (CICR), sie bestreitet den
Großteil der elektromechanischen Kopplung.
Herzmuskelzellen kontrahieren, wenn in ihrem Zytosol der Ca++-Spiegel auf >100 nM ansteigt. Zu einem Viertel beruht dieser Anstieg auf dem Einstrom von Ca++ durch die erregte Zellmembran, zu ~75% stammt er aus dem intrazellulären Speicher: Das Aktionspotential öffnet am Ende von transversalen (T-) Tubuli Ca++-Kanäle in der Nähe des sarkoplasmatischen Retikulums. An dessen Membran regt der Ca++-Einstrom aus den T-Tubuli Calciumkanäle (Ryanodinrezeptoren, RyR) in der Membran des
sarkoplasmatische Retikulums an. Diese Koppelung nennt man
calciuminduzierte Calciumfreisetzung (CICR), sie bestreitet den
Großteil der elektromechanischen Kopplung.
Ventrikuläre
Kardiomyozyten sind sehr klein: Sie haben eine Länge von 1/10 mm und
einen Durchmesser von etwa 20 µm. Im Bereich der Glanzstreifen (disci intercalares) sind sie untereinander zu einem funktionellen Synzytium
verbunden - über gap junctions, an denen Aktionspotentiale von erregten auf unerregte Herzmuskelzellen überspringen.
Auf diese Weise nehmen normalerweise sämtliche Myozyten des Herzmuskels
an Kontraktionen teil, jeder Herzschlag erreicht im
muskelphysiologischen Sinne automatisch ein "Maximum".


Abbildung: Herzmuskelgewebe
Nach einer Vorlage in Boron / Boulpaep: Concise Medical Physiology, Elsevier 2021
Mechanische Verknüpfung erfolgt über Desmosomen, elektrische über gap junctions
(hier jeweils durch nur ein Konnexon angedeutet). Über diese
interzellulären Stellen geringen elektrischen Widerstandes kann sich
Strom über den Intrazellulärraum mehrerer benachbarter Myozyten (von
erregten Zellen ausgehend) ausbreiten und noch unerregte Zellen
aktivieren (funktionelles Synzytium). Über den Extrazellulärraum
schließt sich der Stromkreis.
Kontaktstrukturen sind an Glanzstreifen (intercalated discs) konzentriert
 Aktivierung: Aktionspotentiale breiten sich über das Muskelgewebe aus - in
jeder einzelnen Zelle
längs über die Zellmembran (Sarkolemm) und
von hier quer in schlauchförmige Vertiefungen, die transversalen
oder T-Tubuli. Dadurch gelangt die Erregung unmittelbar in die Tiefe
der Zellen.
Aktivierung: Aktionspotentiale breiten sich über das Muskelgewebe aus - in
jeder einzelnen Zelle
längs über die Zellmembran (Sarkolemm) und
von hier quer in schlauchförmige Vertiefungen, die transversalen
oder T-Tubuli. Dadurch gelangt die Erregung unmittelbar in die Tiefe
der Zellen.
Die T-Tubuli nähern sich in der Tiefe der Herzmuskelzelle bis auf 15 nm (0,015 µm) an das - Calciumionen enthaltende - endo- (sarko-) plasmatische Retikulum (SR) an, das wegen dieser Nähe als junktionales sarkoplasmatische Retikulum bezeichnet wird und mit diesem sogenannte Diaden bildet.
Als Diaden bezeichnet man - jeweils aus einem Riesenprotein aufgebaute - brückenartige Verbindungsstrukturen
zwischen dem sarkoplasmatischen Retikulum (Terminalzisternen) und
transversalen (T-) Tubuli einer Herzmuskelzelle ( Abbildung unten). Sie fungieren als Calciumkanäle - man bezeichnet sie als Ca++ release channels (was ihre Funktion beschreibt) oder - weil sie das Alkaloid Ryanodin binden - als Ryanodinrezeptoren
Abbildung unten). Sie fungieren als Calciumkanäle - man bezeichnet sie als Ca++ release channels (was ihre Funktion beschreibt) oder - weil sie das Alkaloid Ryanodin binden - als Ryanodinrezeptoren (Ryr - im Herzen liegt die Isoform Ryr2 vor). Jede ihrer vier Untereinheiten hat
6 membrandurchspannende α-Helices und eine in die Membran liegende
Halbschleife (loop). Zusammen mit IP3-Rezeptoren gehören sie zum Calcium release-Kanaltypus. So beteiligen sie sich an der elektromechanischen Kopplung.
(Ryr - im Herzen liegt die Isoform Ryr2 vor). Jede ihrer vier Untereinheiten hat
6 membrandurchspannende α-Helices und eine in die Membran liegende
Halbschleife (loop). Zusammen mit IP3-Rezeptoren gehören sie zum Calcium release-Kanaltypus. So beteiligen sie sich an der elektromechanischen Kopplung.
Während der Plateauphase des Aktionspotentials (für ~100
ms) gelangen Calciumionen (Ca++)
über L-Typ-Calciumkanäle vom Extrazellulärraum in die Zelle, was über
sarkoplasmatische Ryanodinrezeptoren die Freisetzung von hier
gespeicherten Calciumionen triggert (die sarkoplasmatisch gespeicherten
Calciumionen sind locker an das Speicherprotein Calsequestrin angelagert). Diese "Calcium-Explosion" wird auch als calciuminduzierte Calciumfreisetzung (CICR: calcium-induced calcium release) bezeichnet. [Ca++] in Sarkoplasma steigt dabei von 0,1 auf 0,5-2,0 µM (eine normale Systole geht vermutlich mit einer sarkoplasmatischen [Ca++] von etwa 0,55 bis 0,75 µM einher), einiges davon bindet an Troponin C und löst die Kontraktion aus.
Ein kleiner Teil des Calciums
wird von
Mitochondrien aufgenommen, die ATP nachsynthetisieren. (Mitochondrien
nehmen ein Drittel des myokardialen Zellvolumens in Anspruch, was die
enorme Intensität des myokardialen Energiestoffwechsels widerspiegelt.)
Als calciuminduzierte Calciumfreisetzung (CICR: Ca++-induced Ca++ release) bezeichnet man die Freisetzung von Ca++
in das Sarkoplasma der Herzmuskelzelle - aus Speichern des
sarkoplasmatischen Retikulums - in Reaktion auf Calciumionen, die im
Rahmen der Erregung über L-Typ-Calciumkanäle (transversale Tubuli) an
Ryanodinrezeptoren (sarkoplasmatisches Retikulum) gelangen (trigger Ca++).
Man vermutet, dass das Trigger-Calcium eines einzelnen durch ein
Aktionspotential aktivierten tubulären L-Typ Calciumkanals eine Gruppe
von bis zu 20 Ryanodinkanälen (Ca++ release channels) im sarkoplasmatischen Retikulum aktivieren kann.
CICR als effizienter Verstärkungsmechanismus: Über L-Typ Calciumkanäle der Zellmembran in den Myozyten eingedrungenes Ca++
führt zu Freisetzung von Calciumionen aus dem junktionalen sarkoplasmatischen
Retikulum (vgl. weiter unten). Ein einziger solcher CICR-Vorgang kann in einer etwa 1 µm
großen Mikrodomäne des Myozyten den Calciumspiegel im Sarkoplasma ([Ca++]) anheben (calcium spark). Pro Herzmuskelzelle addiert sich der Effekt von etwa 104 sparks pro Erregungs in praktisch synchroner Weise, etwa 50% des im SR gespeicherten Calciums wird dabei freigesetzt. Die einzelnen Mikroeffekte summieren sich zu einem Anstieg der sarkoplasmatischen [Ca++]
- für ca. 50 µs von etwa 0,1 µM auf rund 1 µM -, der länger andauert
als das Aktionspotential (die Calciumfreisetzung aus Ryr-Calciumkanälen
dauert länger an als die von L-Typ Calciumkanälen).
Begrenzte Verstärkung: Im Rahmen der Erregung transversaler Tubuli über L-Typ Calciumkanäle freigesetztes Ca++ bewirkt die Freisetzung von Ca++ aus dem SR via release channels und es bestünde die theoretische Möglichkeit einer potitiven Rückkopplung (unkontrollierten Verstärkung). Dass es im Rahmen der CICR dazu nicht kommt, liegt wohl daran, dass es für die Aktivierung der Ryanodinrezeptoren eines sehr hohen Anstiegs der subsarkolemmalen Calciumkonzentration bedarf (ein flacherer Anstieg der [Ca++] lässt die Ryanodinrezeptoren "unbeeindruckt"). Dieser Anstieg bleibt aber räumlich und zeitlich limitiert (auf jeweils durch einen L-Typ Calciumkanal definierten Cluster angrenzender Ryanodinrezeptoren, in dem eine Art Alles-oder-Nichts-Aktivierung erfolgt - sogenanntes cluster bomb model). Die SR-Calciumkanäle (Ryanodinrezeptoren) werden anschließend innerhalb von 30-100 ms inaktiv.
Sarkoplasmatischer Calciumspeicher: Die Menge an Ca++, das
zu einer gegebenen Zeit im sarkoplasmatischen Retikulum gespeichert
ist, bestimmt die Intensität der Kontraktion unmittelbar nachfolgender
Systolen. Zu den Faktoren, welche diese Speicherung beeinflussen,
gehören
 die extrazelluläre Ca++-Konzentration (die von Organismus sehr genau reguliert wird),
die extrazelluläre Ca++-Konzentration (die von Organismus sehr genau reguliert wird),
das Ausmaß des Einstroms über tubuläre L-Typ Calciumkanäle (verstärkt durch Katecholamine, abgeschwächt durch Calciumkanalblocker),
die Herzfrequenz, die wiederum die Dauer von Systole (Calciumeinstrum aus dem Extrazellulärraum) und Diastole (Herausbefördern von Ca++ aus dem Sarkoplasma) beeinflusst.

Abbildung: Herzmuskelzelle
Nach einer Vorlage in Herring / Paterson, Levick's Introduction to Cardiovascular Physiology, 6th ed. 2018
Sarkomere erstrecken sich jeweils zwischen
zwei Z-Streifen und sind in Serie angeordnet. Sie sind das kontraktile
Element des Muskels.
Transversale Tubuli sind mit dem Extrazellulärraum verbundene
Einstülpungen des Sarkolemms; sie sind parallel zu Z-Streifen angeordnet. Sie tragen spannungsabhängige L-Typ-Ca
++-Kanäle
(Dihydropyridinrezeptoren), durch sie dringt Ca
++ einerseits in das Sarkoplasma ein, andererseits triggert es die Freigabe von Ca
++
aus dem sarkoplasmatischen Retikulum. Bei Herzmuskelzellen sind L-Typ-Ca
++-Kanäle für die elektromechanische Koppelung unbedingt erforderlich.
Die Kontaktstellen mit
Ausläufern des endoplasmatischen Retikulums bezeichnet man als
Diaden.
Hier befinden sich Ryanodinrezeptoren (
rot), das sind
Calciumkanäle
in der Wand des sarkoplasmartischen Retikulums (SR), die Ca
++ aus dem SR in das Sarkoplasma diffundieren lassen - das löst die Kontraktion aus.
Das sarkoplasmatische Retikulum legt sich wie ein Netz über Myofibrillen. Es ist ein Zwischenspeicher für Ca
++-Ionen,
die bei Erregung in das Zytosol freigesetzt werden, zwischen die
Filamente diffundieren und die elektromechanische Koppelung
(Kontraktion) vollenden. Anschließend werden sie wieder in das
Retikulum zurückbefördert (Ca
++-ATPasen, SERCA
), wodurch sich der Muskel wieder entspannen kann.
Mitochondrien nehmen bis zu einem Drittel des Zellvolumens in Anspruch, sie versorgen die Myozyten mit ATP, was die
Energieversorgung
des (niemals ruhenden) Herzens sichert. Die Mitochondrien reihen sich
zwischen den Myofibrillen auf. Die Produktion des ATP erfolgt durch
oxidative Phosphorylierung. Die Energieversorgung ist abhängig von der
koronaren Perfusion

Kontraktionsmechanismus: Depolarisation bewirkt an transversalen Tubuli den Einstrom von Ca++-Ionen durch spannungsabhängige L-Typ-Calciumkanäle (auch Dihydropyridinrezeptoren DHPR - bezeichnet wegen seiner Affinität zum Calc
iumkanalblocker DHP) in die Zelle. Dabei steigt [Ca++] im Sarkoplasma an, von 0,1 auf bis zu 2 mM. Einige dieser Calc
iumionen binden an Troponinmoleküle der kontraktilen Filamente und lösen die elektromechanische Kopplung (Auslösung einer Kontraktion infolge Erregung der Muskelzelle) aus.

Abbildung: Zeitverlauf von Aktionspotential, Calciumkonzentration und Kontraktion einer Herzmuskelzelle
Die Depolarisierung triggert die Erhöhung der intrazellulären Calciumkonzentration, und diese die Kontraktion. Der Ca++-Zeitverlauf erfolgt gegen die Depolarisationskurve zeitverschoben, die Kontraktion noch später
 Über den Kontraktionsmechanismus in quergestreiften Muskelzellen s. dort
Über den Kontraktionsmechanismus in quergestreiften Muskelzellen s. dort

Abbildung: Calciumtransport und Mechanismen der Modulierung
Nach Niggli E, Ullrich ND, Gutierrez D, Kyrychenko S,
Polakova E, Shirokova N. Posttranslational modifications of cardiac
ryanodine receptors: Ca2+ signaling and EC-coupling. BBA Mol Res 2013; 1833: 866-75
Die Modulierung kann über Calciumionen und Phosphorylierung erfolgen. LTCC (L-type calcium channels) aktivieren RyR (Ryanodinrezeptoren) und regen Calciumeinstrom aus dem sarkoplasmatischen Retikulum (SR) an (CICR: calcium-induced calcium release).
Das Ausmaß der Phosphorylierung der RyR bestimmt deren Empfindlichkeit gegenüber Ca++: Aktivierung von ß1-Adrenozeptoren (ß-AR) aktiviert Adenylatzyklase (AC)
und Proteinkinase A (PKA). PKA phosphoryliert RyR und Phospholamban
(PLB) und regt dadurch den Calciumdurchsatz des Kardiomyozyten an.
Nach CICR und Kontraktion wird Ca++ in das SR zurückgepumpt: Erhöhter zytoplasmatischer Calciumspiegel aktiviert die Ca++/calmodulinabhängige Proteinkinase II (CaMKII): CaMKII hilft
den Calciumspiegel im Sarkoplasma zu senken - durch Inhibition von LTCC und RyR sowie Aktivierung der SERCA (Sarcoplasmic / endoplasmic reticulum calcium ATPase) durch Inhibition des PLB.
SERCA an der Membran des sarkoplasmatischen Retikulums, sowie (hier nicht gezeigt) Na/Ca-Austauscher und Calciumpumpen in der Außenmembran (Sarkolemm) regulieren den
sarkoplasmatischen Calciumspiegel
 Der Calciumspiegel steigt im Sarkoplasma
einer erregten Herzmuskelzelle auf 0,5-2 µM an (im Skelettmuskel bis
auf 10 µM), so wird nur ein Bruchteil der vorhandenen
Aktin-Myosin-Querbrücken aktiviert (im Skelettmuskel ist die
Aktivierung komplett). Das bedeutet, die Kontraktion ist submaximal
(meist um die 40% des Maximums). Erhöhung des systolischen
Calciumanstiegs (z.B. durch Adrenalin) steigert auch die Zahl der
aktiven Querbrücken und damit die Schlagkraft.
Der Calciumspiegel steigt im Sarkoplasma
einer erregten Herzmuskelzelle auf 0,5-2 µM an (im Skelettmuskel bis
auf 10 µM), so wird nur ein Bruchteil der vorhandenen
Aktin-Myosin-Querbrücken aktiviert (im Skelettmuskel ist die
Aktivierung komplett). Das bedeutet, die Kontraktion ist submaximal
(meist um die 40% des Maximums). Erhöhung des systolischen
Calciumanstiegs (z.B. durch Adrenalin) steigert auch die Zahl der
aktiven Querbrücken und damit die Schlagkraft.
Das sarkoplasmatische Retikulum
(SR) - ein Netzwerk 20-60 nm weiter intrazellulärer Tubuli, die
Myofibrillen strumpfartig umgeben und zahlreiche Kontaktstellen mit transversalen Tubuli aufweisen - konstituiert den intrazellulären Ca++-Speicher (gebunden an Calsequestrin) und nimmt ~5% des Zellvolumens in Anspruch.
Es bildet zwei miteinander verbundene Kompartimente:
 Ein sich über Myofibrillen erstreckendes Netzwerk (network SR), das via (durch Phospholamban gesteuerte) Ca++-Pumpen (SERCA) aus dem Zytosol freigesetzte Ca++-Ionen aufnimmt; und
Ein sich über Myofibrillen erstreckendes Netzwerk (network SR), das via (durch Phospholamban gesteuerte) Ca++-Pumpen (SERCA) aus dem Zytosol freigesetzte Ca++-Ionen aufnimmt; und
ein junktionales (junctional SR), das Ca++ speichert, enge Kontaktstellen mit dem T-System (Diaden) aufweist und - bei dessen Depolarisierung - Ca++ in die Zelle abgibt. An der Kontaktstelle sitzt ein Riesenprotein (2,3 MDa), das als Ryanodinrezeptor (Ryr), Ca++ release channel oder calcium-induced calcium release (CICR) channel bezeichnet wird.
 ~80% (75-90) des während eines Kontraktionszyklus in das Sarkoplasma gelangten Ca++ stammt aus dem sarkoplasmatischen
Retikulum; ~20% (10-25) kommen aus dem Extrazellulärraum. Extrazelluläres Ca++ - das über L-Typ Calciumkanäle in den Myozyt einströmt - ist dennoch von entscheidender Bedeutung, denn es triggert die Freisetzung von Ca++ aus dem sarkoplasmatischen Retikulum (über Ryanodinrezeptoren - CICR).
~80% (75-90) des während eines Kontraktionszyklus in das Sarkoplasma gelangten Ca++ stammt aus dem sarkoplasmatischen
Retikulum; ~20% (10-25) kommen aus dem Extrazellulärraum. Extrazelluläres Ca++ - das über L-Typ Calciumkanäle in den Myozyt einströmt - ist dennoch von entscheidender Bedeutung, denn es triggert die Freisetzung von Ca++ aus dem sarkoplasmatischen Retikulum (über Ryanodinrezeptoren - CICR).

Abbildung: Elektromechanische Kopplung in einer Herzmuskelzelle
Nach Knollmann BC, Roden DM, A genetic framework for improving arrhythmia therapy. Nature 2008; 451: 929-36
Grüne Pfeile: Transmembranaler Calc
iumstrom.
Junctin und Triadin sind Proteine in der Wand des sarkoplasmatischen Retikulums, die an der calciuminduzierten Calc
iumfreisetzung beteiligt sind.
Sarkoglykan verbindet das Zytoskelett mit der extrazellulären Matrix.
 Proteine, deren Gene bei primärer Arrhythmie (Herzrhythmusstörung) mutiert sein können (Erregungsstörung)
Proteine, deren Gene bei primärer Arrhythmie (Herzrhythmusstörung) mutiert sein können (Erregungsstörung)
 Proteine, deren Gene bei Kardiomyopathien mutiert sein
können, was ebenfalls zu Arrhythmien führen kann
Proteine, deren Gene bei Kardiomyopathien mutiert sein
können, was ebenfalls zu Arrhythmien führen kann
 Triadin
hat Einfluss auf die Calciumfreisetzung aus dem sarkoplasmatischen
Retikulum, indem es mit dem Ryanodinrezeptor interagiert. Es fungiert
als Ca++-Sensor und beeinflusst die Interaktion zwischen dem Ryanodinrezeptor und Calsequestrin.
Triadin
hat Einfluss auf die Calciumfreisetzung aus dem sarkoplasmatischen
Retikulum, indem es mit dem Ryanodinrezeptor interagiert. Es fungiert
als Ca++-Sensor und beeinflusst die Interaktion zwischen dem Ryanodinrezeptor und Calsequestrin.
Das in der Membran des sarkoplasmatischen Retikulums verankerte Protein Phospholamban (PLN oder PLB) ("Phosphatfänger") bremst im dephosphorylierten (aktiven) Zustand die Aktivität der SERCA (Ca++-Pumpe) und damit die Ca++-Aufnahme in das
sarkoplasmatische Retikulum (Abbildung oben).
Phosphorylierung des Phospholambans schwächt dessen Bremswirkung auf den Calcium-Einwärtstransport ab und beschleunigt die Relaxation. Die Phosphorylierung erfolgt durch cAMP-abhängige Proteinkinase A (PKA) - insbesondere unter der Wirkung von Adrenalin / Noradrenalin, die PKA aktivieren und so die Relaxation beschleunigen (positive Lusitropie).
Dephosphorylierung
des PLN stellt seine Hemmwirkung auf SERCA wieder her, sie kann durch
das Holoenzym (regulatorische + katalytische Untereinheit) Typ 1-
Protein Phosphatase (PP1) erfolgen. PP1 ist ein bedeutender Regulator des Calciumumsatzes und der Reaktion des Herzmuskels auf ß1-adrenerge
Stimulation. Ist die PP1-Aktivität (die durch endogene Inhibitoren
reguliert wird) zu hoch, schwächt das die Kontraktionsfähigkeit der
Zelle.
Calsequestrin befindet sich im junktionalen sarkoplasmatischen Retikulum ( Abbildung unten) und kann eine hohe Zahl an Calciumionen binden.
Desmosomen dienen der mechanischen Verknüpfung von Herzmuskelzellen.
Connexinkanäle verbinden
benachbarte Myozyten in gap junctions, sodass elektrische
Potentialänderungen direkt von Zelle zu Zelle übertragen werden können.
Sarcoglykane verknüpfen das Zytoskelett der Muskelzelle mit der extrazellulären Matrix und dienen so der mechanischen Verankerung.
In der ruhenden Herzmuskelzelle beträgt die Konzentration an freien Calciumionen ~10-7M. Bei Erregung steigt sie bis 10-5M, also etwa 100-fach an. (Im Extrazellulärraum beträgt [Ca++] ~10-3M.)
Zeitlich-räumliche Optimierung: Ca++ bindet an Troponin C.
Durch die räumliche Anordnung der Permeasen werden in koordinierter
Weise gleichzeitig oberflächliche und tiefer gelegene Teile der
Muskelfaser mit Ca++ "überflutet", was zu hoher Effizienz des Kontraktionsvorgangs führt.
Der weitere Vorgang erfolgt analog dem Kontraktionsmechanismus in der Skelettmuskulatur.
Ca++-Ionen verlassen anschließend die Herzmuskelzelle aktiv (Ca-ATPase) oder im Austausch gegen Natrium (NCX: Na/Ca-Austauscher). Der NCX arbeitet ladungsabhängig: Im nicht
erregten Myozyt (diastolisch) fördert er Ca++ aus der Zelle; im
erregten (systolisch) in die Zelle. Insgesamt überwiegt beim NCX der
Netto-Auswärtstransport von Ca++.
Vergleich der elektromechanischen Kopplung bei quergestreiften und glatten Muskelzellen s. dort
Die postsystolische Entspannungsfähigkeit nennt man Lusitropie
Herzqualitäten s. dort
Um die
Herzmuskelzelle wieder zu entspannen, muß die sarkoplasmatische
Calciumkonzentration sinken. Dadurch dissoziieren Calciumionen von
Troponin C und verlieren so ihre Wirkung auf das kontraktile System.

Abbildung: Calcium und Herzmuskelaktivität
Nach einer Vorlage in Herring / Paterson, Levick's Introduction to Cardiovascular Physiology, 6th ed. 2018
Links: Der Einstrom von Ca++ aus dem Extrazellulärraum über L-Typ-Calciumkanäle trägt rund 20% (10-25%) zur elektromechanischen Kopplung bei und triggert die Öffnung von Calciumkanälen des sarkoplasmatischen Retikulums (CICR: calciuminduzierte Calciumfreisetzung). Etwa 80% (75-90%) des für die elektromechanische Kopplung benötigten Calciums stammt aus dem sarkoplasmatischen Retikulum.
Rechts: Unter Mitwirkung von Phospholamban wird Ca++ in der Diastole wieder aufgenommen und z.T. an Calsequestrin gebunden. Calcium verlässt die Herzmuskelzelle über Austausch mit Natrium (1 Ca++ gegen 3 Na+, netto eine positive Ladung in die Zelle - Beitrag zur Plateauphase des Aktionspotentials), sowie direkt energieverbrauchend
Das sarkoplasmatische Retikulum (SR) nimmt 5% des Volumens der Herzmuskelzellen in Anspruch. Das junktionale SR enthält Ca++ in relativ hoher Konzentration (etwa 1 mM) und ist dicht mit Ryanodinrezeptoren ausgestattet, seine Funktion ist die Freisetzung von Ca++ für die elektromechanische Kopplung. Die Ryanodinrezeptoren (calcium release channels) sind riesige T-förmige Permeasen (etwa 2300 kDa) mit fußförmigen Calciumkanälen, die nur wenige Nanometer von der Membran transversaler Tubuli entfernt enden (auf einen tubulären L-Typ Calciumkanal kommen etwa 10 SR-Ryanodinrezeptoren).
Während das junktionale SR auf die rasche Freisetzung von Calciumionen spezialisiert ist, besteht die Aufgabe des "Netzwerk-SR" in der (ATP-betriebenen) Rückgewinnung von (während der Kontraktion in das Sarkoplasma gelangten) Calciumionen ( Abbildung).
In der Relaxationsphase (restitution) wird Ca++ in intrazelluläre Speicher (75-90%) sowie in den
Extrazellulärraum (10-25%) verbracht, die Calc
iumkonzentration im Zytoplasma nimmt ab. Das erfolgt über mehrere Wege:
Transport von Ca++ in den Extrazellulärraum - über Na/Ca-Austauscher NCX1 (für höhere [Ca++]-Werte im Sarkoplasma) und Ca-Pumpen (PMCA: Plasma membrane Ca ATPase, kardiale Subtypen 1, 2 und 4 - arbeiten auch bei niedrigen [Ca++]-Werten im Sarkoplasma)
Transport von Ca++ in das sarkoplasmatische Retikulum (durch SERCA2a). SERCA wird reguliert durch Phospholamban (s. oben)
Aufnahme von Calciumionen in
Mitochondrien: Die innere Mitochondrienmembran verfügt über selektive Calciumkanäte (MiCas)
Die als Lusitropie bezeichnete Eigenschaft bezieht sich auf myokardiale Entspannung.
Da Calciumionen aktiv aus dem Sarkoplasma entfernt werden müssen, um
die Zelle "weich" zu machen, ist die diastolische Relaxation
ATP-abhängig.
Positive Lusitropie:
Phosphorylierung des Phospholambans durch
Proteinkinase A (PKA - Anregung durch cAMP) beschleunigt die Rückführung von
Ca
++ ins endoplasmatische Retikulum.
Dies erklärt den positiv lusitropen Effekt
einer
ß1-Rezeptor-Stimulierung (
erhöht die PKA-Aktivität).
Sympathikusaktivität fördert die diastolische Entspannung des Myokard.
Noradrenalin bewirkt Phosphorylierung von Phospholamban, und da dieses
die Aufnahme von Ca++ in das sarkoplasmatische Retikulum fördert, relaxiert das Myokard rascher. Allerdings ist auch der Wiederaustritt von Ca++
ins Zytosol erleichtert, wenn die Herzmuskelzelle wieder erregt wird
(positiv inotroper Effekt). Dieser Vorgang spielt sich am
sarkoplasmatischen Retikulum ab; ohne Katecholaminwirkung hätte ein
vermehrter Ca++-Einstrom aus dem Extrazellulärraum einen negativ lusitropen Effekt.
Negative Lusitropie: Die Relaxation des Herzmuskels wird durch folgende Faktoren behindert:
Calciumüberladung des Zytosols

verminderte Funktion der
Calciumpumpen: Ca
++-ATPase, Na
+-Ca
++-Austauscher an der Zellmembran
verminderte Funktion der SERCA (sarkoplasmatische Ca
++-ATPase)
Im sarkoplasmatischen Retikulum werden Calciumionen schwach und daher reversibel, aber mit hoher Kapazität - 65 Ca++
pro Molekül Calsequestrin - gebunden.
Mechanismus und Steuerung der Kontraktion
Alle Fasern des Herzmuskels sind über gap junctions elektrisch miteinander verbunden; Erregungswellen
breiten sich über sämtliche Kardiomyozyten aus. Alle Fasern nehmen an der Systole teil, daher ist jeder Herzschlag ein "Maximum", das von einer
völligen Entspannung gefolgt ist.
Allerdings sind bei ruhiger Herzaktion nur etwa 40% der Aktin-Myosin-Querbrücken aktiv. Bei körperlicher Belastung bzw. allgemein unter Stressbedingungen steigt dieser Anteil, die Systole wird kräftiger (positive Inotropie). Das ist durch zwei Mechanismen verursacht:
Calciummenge:
Katecholamine steigern
das Ausmaß des Ca++-Einstroms durch L-Typ-Ca++-Kanäle (Dihydropyridinrezeptoren). Über Aktivierung von ß1-Rezeptoren steigt [cAMP], es kommt zu Aktivierung der Proteinkinase A und (Phosphorylierung) von spannungsabhängigen Ca++-Kanälen, verstärkten Ca++-Einstrom und erhöhtes [Ca++] für die elektromechanische Kopplung (Troponin)
Calciumempfindlichkeit: Mit der
diastolischen Füllung steigt die Kontraktionskraft (
Frank-Starling-Mechanismus), weil
die Sensitivität der Myofibrillen gegenüber Ca
++-Ionen mit zunehmender Dehnung zunimmt.
Lage der Kardiomyozyten und Expression von Protein-Isoformen: Lokale Unterschiede (subepi- vs. subendokardial gelegene Schichten) des Funktionsprofils von
Myokardzellen sind durch unterschiedliche Isoform-Expression
kontraktiler und regulatorischer Proteine bedingt.
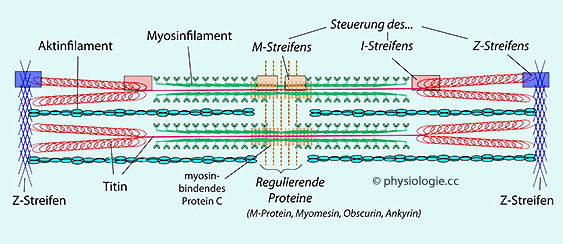
Abbildung: Organisation eines myokardialen Sarkomers
Nach Katz AM, Zile MR, New molecular mechanism in diastolic heart failure. Circulation 2006; 113: 1922-5
Das 1,8-2,0 µm lange Sarkomer ist die Region zwischen zwei Z- (Zwischen-) Streifen. An ihnen setzen Aktin- und Titinfilamente an.
Aktinfilamente sind ~1 µm lang und 6 nm dick. Zusammen mit Titin füllen sie den isotropen (I-) Streifen. Im anisotropen (A-) Streifen interagieren sie mit Myosin, was den Mechanismus der Kontraktion unterstützt.
Die 150 nm langen Myosinmoleküle
sind golfschlägerförmig; ihr Kopfteil ist bei der Kontraktion aktiv.
Myosinfilamente sind 1,6 µm lang, messen 11 nm im Durchmesser und
bestehen aus ~400 Myosinmolekülen.
Die
aktinfreie H- ("helle") Zone in der Mitte des Sarkomers beinhaltet M-
("Mittelscheiben") Streifen. Diese enthalten regulierende Proteine wie
z.B. Myomesin, das mit Titin interagiert. Titin verleiht strukturelle Festigkeit und bestimmt (zusammen mit Kollagen im extrazellulären Raum) die passive Dehnbarkeit
des Herzmuskels. In der (Myosinfilamente enthaltenden) A-Zone ist Titin
ziemlich rigide, in der (äußeren, nur Actinfilamente enthaltenden)
I-Zone elastischer. Titin interagiert mit mehreren Proteinen, darunter
M-Protein, Myomesin, Onscurin und Ankyrin. Ankyrine verankern Ionenkanäle in der Zellmembran, myosinbindende Proteine stabilisieren das Sarkomer, Obscurin ist ein großes Signalprotein.
Die Rechtecke in der Abbildung markieren Zonen, in denen Titin an
Signaltransduktion beteiligt ist (Bereiche des M-, I- und Z-Streifens)
Über das Skelettmuskelsarkomer s.
dort
Titinmoleküle
erstrecken
sich über das gesamte Sarkomer; in
der Sarkomermitte sind sie über das
myosinbindende Protein (orangefarben in der Abbildung) an Myosin
fixiert. Titin wirkt wie eine Feder, die gedehntes Myokard wieder zu
verkürzen hilft. Diese Gegenkraft beginnt bei einer Sarkomerlänge von
etwa 2 µm und nimmt bei weiterer Dehnung steil zu.
Im Herzmuskel
finden sich zwei Teilsequenzen im I-Streifen des Titinmoleküls (N2-B
und N2-A), die sich in ihrer Dehnbarkeit
unterscheiden und individuell unterschiedlich stark ausgeprägt sind.
Regulierende
Proteine (Ankyrin etc) interagieren mit Titin im Bereich des
M-Streifens.
Entlang des Titinmoleküls wechseln mechanische
Eigenschaften: Elastisch im Bereich des I-Streifens, rigide im Bereich
der A-Zone. Zonen für Signaltransduktion im M-, I- und
Z-Streifenbereich sind in der Abbildung angezeigt.
Titin bestimmt die Dehnbarkeit (Compliance)
der Sarkomere bzw. des Myokards; sie sind zu den kontraktilen
Filamenten parallelgeschaltet, diese sind aber im diastolischen Zustand
fast widerstandlos verschiebbar. Normale Myokardzellen werden in vivo nicht über ~2,3 µm lang; intensivere Streckung resultiert in geringer Dehnbarkeit, die Herzmuskelzelle "versteift".
Mutiertes "Riesentitin" ermöglicht
eine Dehnung der Sarkomere bis ~4 µm.
 Abbildung: Phasen des Kontraktionsablaufs
Abbildung: Phasen des Kontraktionsablaufs
Nach einer Vorlage in Herring / Paterson, Levick's Introduction to Cardiovascular Physiology, 6th ed. 2018
Oben: Ruhezustand. Tropomyosin blockiert die Bindungsstellen am Aktin (gelber Stern).
Mitte: Freigesetzte
Calciumionen verändern die Position des Troponinkomplexes, die
Bindungsstellen für Myosinköpfe werden frei für die Querbrückenbildung.
Unten: Verwinkelung des
Myosinkopfes (mit gebundenem Aktin) führt zu einer "Ruderbewegung", der
Z-Streifen rückt um eine Strecke von 5-10 nm zur Sarkomermitte.
Anschließend löst sich der Myosinkopf vom Aktinfilament und kippt -
unter ATP-Verbrauch - in
die Ausgangslage zurück, um neuerdings einen Aktin-Bindungspartner zu
engagieren. Die Energie für den nächsten Kraftschlag wird also in das
Zurückklappen des Myosinkopfs (ähnlich wie in das Spannen einer Feder)
investiert.
Insgesamt verfügt ein Myosinfilament über etwa 400 Myosinköpfe
Die Abbildung
zeigt ein Schema des Kontraktionsablaufs in einer
Herzmuskelzelle. Er ist sehr ähnlich demjenigen in einer
Skelettmuskelzelle.
Jeder einzelne Myosinkopf fungiert als unabhängiger Kraftgenerator, und
die (nicht synchron ablaufende) Aktion von hunderten davon (pro
Myosinfilament) summiert sich (solange die Bindungsstellen an den
jeweils 6 umliegenden Aktinfilamenten unter Ca++-Einwirkung
auf Troponin frei sind) zu einem kontinuierlichen Zug an den
Z-Streifen. Diese bewegen sich (soferne die Gegenkraft nicht überwiegt)
zur Sarkomermitte und verkürzen so
die Muskelfaser insgesamt. Die Addition dieses Effekts an Muskelzellen
im myokardialen Synzytium ergibt schließlich die systolische
Kontraktion, der Druck im Vorhof / Ventrikel nimmt zu, Blut wird
entsprechend den Gesetzen der Herzmechanik weiterbefördert.
Die Geschwindigkeit des zyklischen Ablaufs der Querbrückenbildung / Querbrückenlösung (crossbridge cycling) - und damit des Aneinander-vorbei-Gleitens der Filamente im Sarkomer - hängt von der Isoform der Myosin-Schwerketten (MHC: myosin heavy chain) ab. Der Herzmuskel erwachsener Menschen hat 97% langsam gleitendes ß-MHC (die führende Isoform im Herzmuskel des Menschen) und 3% rasch gleitendes α-MHC (vor allem in den Atrien exprimiert).
Troponin (TNN) bindet Calciumionen und reguliert die Kontraktion. Ca++-Ionen binden an Troponin C,
was die räumliche Struktur des Troponinkomplexes ändert (Tropomyosin
gibt die Reaktionsstellen für Myosinköpfe frei). Der Troponinkomplex besteht aus drei Untereinheiten:
Troponin C bindet Calciumionen (die im Rahmen der elektromechanischen Kopplung in das Sarkoplasma freigesetzt wurden)
Troponin I
wirkt inhibitorisch
auf die Bindung des Myosinkopfes an Aktinmoleküle (verhindert die
Kontraktion), bei Calciumanlagerung an Troponin C verliert es seine
inhibitorische Wirkung
Troponin T bindet den Komplex an Tropomyosin und unterstützt die Positionierung an das Aktin.
Die im
Herzen vorliegende Isoform des Troponins ist Troponin C1 (TNNC1). Der Ca++-TNNC1-Komplex hebt anschließend die Hemmung der kardialen Isoform des Troponin I (TNNI3) am Aktin auf.
Troponin wird bei Beschädigungen von Muskelzellen vermehrt in den Kreislauf freigesetzt. Kardiales Troponin I und Troponin T sind Marker für Hermuskelnekrosen.
Tropomyosin
(TPM) ist ein 43 nm langes, bandförmiges regulatorisches Protein, das sich zwischen die
beiden Ketten der Aktinmoleküle legt, dabei sieben G-Aktin-Untereinheiten abdeckt und an einem Ende einen
Troponinkomplex trägt. Die Ca++-bedingte Aufhebung
der Hemmung des Troponin I (TNNI3) am Aktin bewirkt das Wegkippen des
Tropomyosins (TPM1) vom Troponin T2 (TNNT2), und die Myosinköpfe können
mit Aktinmolekülen in Interaktion treten - der Muskel kontrahiert.
Die höchste Kontraktionskraft liefert eine Myokardzelle bei einer Sarkomerlänge von 2,2 µm, also bei starker Vordehnung. Bei
einem normalen enddiastolischen Volumen beläuft sich die Sarkomerlänge
auf 1,8-2,0 µm; das bedeutet, das Myokard arbeitet im ansteigenden Teil der Kraft-Volumen-Kurve (die Kraftausbeute nimmt mit der diastolischen Füllung zu - in Übereinstimmung mit der Aussage des Frank-Starling-Mechanismus).
Übertragung der Kontraktionskraft über das Netz an Kardiomyozyten: Jede einzelne Muskelzelle trägt mit ihrer Kontraktion zur systolischen Druckentwicklung bei. Das bedeutet, dass die Zellen untereinander
mittels stabiler mechanischer Verknüpfungen verbunden sein müssen. Diese gegenseitige Verankerung erfolgt
mittels komplexer Molekülnetze
(Zellkontakte, junctional complexes) - zwischen Myozyten via Desmosomen (maculae adhaerentes), mit anderen Zellen mittels Hemidesmosomen. Cadherine spielen für solche Kraftübertragungsstellen eine besondere Rolle.
Ruhedehnungskurve, Druck-Volumen-Beziehung
Ähnlich wie beim Skelettmuskel, können auch beim Herzmuskel Beziehungen zwischen Vordehnung und Kontraktionskraft
quantifiziert werden. Sofern nicht ein isolierter Muskelstreifen,
sondern das ganze Organ Gegenstand der Untersuchung ist, werden statt
der Vordehnung (L) der Ventrikelinhalt (L3), und statt der Kraft (K) der Druck im Ventrikel (K/L2) gemessen und gegeneinander aufgetragen.
So ergeben sich Druck-Volumen-Diagramme,
und da Druck mal Volumen die Dimension Arbeit (K mal L) hat, können die
Flächen im Diagramm, die bei einem Herzschlag umstrichen werden, als
Maß für die aufgebrachte mechanische Arbeit des Ventrikels während
einer Systole herangezogen werden.

Abbildung: Ruhedehnungskurve (grün), U-Maxima (rote Linie)
Nach einer Vorlage bei adinstruments.com
Ein Herzschlag überstreicht
die grün schraffierte Druck-Volumen-Kurve, deren Fläche ein Maß für die
geleistete Arbeit während der Kontraktion ist. Alle Systolen enden auf einer Geraden der jeweils gültigen U-Maxima (rot).
RDK = Ruhedehnungslurve (grün)
Näheres s. dort
Eine Kontraktion, an der alle verfügbaren Muskelfasern teilnehmen, führt zu einem sogenannten Maximum.
Jeder Herzschlag erreicht (am Ende der Systole) automatisch ein
Maximum, da ja sämtliche Muskelfasern an der Kontraktion teilnehmen
(daher nennt man einen solchen zusammenhängenden Zellverband ein funktionelles Synzytium).
Eine
Kontraktion, die (wie ein normaler Herzschlag) aus einer
iso(volu)metrischen und dann einer isotonen bzw. auxotonen Phase
besteht, bezeichnet man als Unterstützungszuckung. Deren Maximalwert ist im Druck-Volumen-Diagramm
durch die Lage auf der Linie der U-Maxima (end-systolic pressure-volume relation ESPVR) gekennzeichnet.
Kontraktionen aus unterschiedlicher diastolischer
Vorfüllung (preload) und mit unterschiedlichem Druck im arteriellen Gefäß erreichen unterschiedliche
Maximalpunkte im Druck-Volumen-Diagramm, die auf der sogenannten U-Kurve
bzw. -Gerade (Verbindung aller Unterstützungs-Maxima - in der Abbildung rote Gerade) liegen. Je weiter diese von den
Ausgangspunkten auf der Ruhedehnungskurve (grün) entfernt sind, desto stärker schlägt das Herz
(Abschätzung der Inotropie).
Kraft, Geschwindigkeit, Belastung
Wie bei jedem Muskel, hängt die Geschwindigkeit
der myokardialen Kontraktion von der Belastung ab ( Abbildung). Ist diese
vernachlässigbar gering, kontrahiert der Muskel mit maximal möglicher
Geschwindigkeit. Mit zunehmender Gegenkraft nimmt die
Verkürzungsgeschwindigkeit ab, was die Entleerung des betreffenden
Herzraumes verlangsamt und die Transportfunktion des Herzens
beeinträchtigen kann.
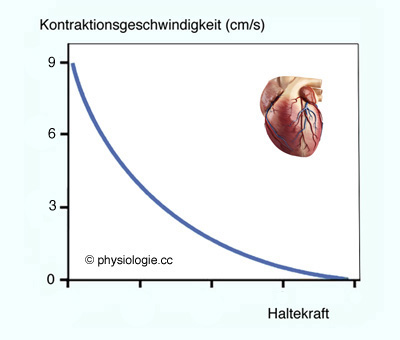
Abbildung: Kontraktionsgeschwindigkeit als Funktion der Belastung
Nach einer Vorlage bei Klabunde RE, Cardiovascular Physiology Concepts. cvphysiology.com
Die
Kontraktionsgeschwindigkeit ist je nach Region des untersuchten
Herzmuskels sehr unterschiedlich. Die Muskelfasern ziehen in anatomisch
komplexer Anordnung durch das Myokard und verkürzen sich entsprechend
lokalen Anforderungen.
Hohe Vorlast verschiebt die
Kurve zu höherer, niedriges zu geringerer Kraftentfaltung (Abszisse:
"Haltekraft"); die maximale Kontraktionsgeschwindigkeit bleibt gleich
(Ordinate).
Positiv inotrope Einflüsse verschieben die gesamte Kurve nach oben und rechts, negativ inotrope nach unten und links
Anders ausgedrückt: Mit zunehmender Nachlast (afterload) nimmt - ceteris paribus - die Auswurfgeschwindigkeit ab.
Die Geschwindigkeit, mit der der Ventrikel kontrahiert, nimmt vor allem mit zwei Faktoren zu:
Wenn die Länge der Muskelfasern zunimmt (also mit steigendem Füllungsvolumen bzw. höherer Vorlast), und
m
it sinkender Gegenkraft (arteriellem Druck bzw. Nachlast).
Positiv inotrop wirkende Substanzen bewirken, dass das Herz bei
gleichbleibender Kontraktionsgeschwindigkeit eine größere Last
bewältigt, bzw. eine gleichbleibende Last mit höherer Geschwindigkeit.
Herzbelastung und Hypertrophie: Über längere Zeit hat der Betrag der Nachlast einen trophischen Effekt: Je höher, desto intensiver ist das "Training" für den Herzmuskel und umso höher wird die Masse des betreffenden Myokards.
So kommt es z.B. bei Behinderungen der diastolischen Blutströmung durch
die AV-Ebene (Klappenstenose) oder systolischen Rückstrom durch eine
undichte AV-Klappe (Klappeninsuffizienz) zu verstärkter Belastung des atrialen Myokards und Vorhofhypertrophie.
Erhöhte Pumpanforderung an die Ventrikel durch wiederholtes intensives körperliches Training (z.B. Radsport) bedingt ein "Sportlerherz",
das Ventrikelmyokard nimmt von ~300 g bis zu ~500 g zu (das führt
u.a. auch zu erhöhtem parasympathischen Einfluss - Ruhe-Bradykardie bis
unter 40 bpm).
Taschenklappenprobleme (systolisch: Stenose, diastolisch: Insuffizienz) führen zu erhöhter Belastung und ebenfalls zu ventrikulärer Hypertophie.
Nimmt die mittlere Belastung wieder ab, sinkt auch der trophische
Effekt und die Muskelmasse reduziert sich allmählich in Richtung "Kontrollwert" zurück.
 "Herzglykoside"
(z.B. Digoxin) reduzieren die Aktivität der Na/K-Pumpe in der Membran (Sarkolemm) der
Kardiomyozyten (3 in der Abbildung) bis um 25%. Dadurch nimmt die Na+-Konzentration
in der Zelle zu und der treibende Natriumgradient für den
Na/Ca-Austauscher ab, was den Auswärtstransport von Ca++ reduziert (Abbildung).
"Herzglykoside"
(z.B. Digoxin) reduzieren die Aktivität der Na/K-Pumpe in der Membran (Sarkolemm) der
Kardiomyozyten (3 in der Abbildung) bis um 25%. Dadurch nimmt die Na+-Konzentration
in der Zelle zu und der treibende Natriumgradient für den
Na/Ca-Austauscher ab, was den Auswärtstransport von Ca++ reduziert (Abbildung).
 Auf diese Weise steigt die intrazelluläre Calciumkonzentration und damit die Kontraktionskraft (positiv inotroper Effekt).
Der Nachteil ist, dass die toxische Dosis nur leicht über der
therapeutischen liegt; Calciumanreicherung im Sarkoplasma kann zu
Nachdepolarisierungen führen, Arrhythmien können auftreten.
Auf diese Weise steigt die intrazelluläre Calciumkonzentration und damit die Kontraktionskraft (positiv inotroper Effekt).
Der Nachteil ist, dass die toxische Dosis nur leicht über der
therapeutischen liegt; Calciumanreicherung im Sarkoplasma kann zu
Nachdepolarisierungen führen, Arrhythmien können auftreten.
Da die Herzglykoside den Rücktransport von Ca++ in das sarkoplasmatische Retikulum nicht beeinflussen, haben sie keinen lusitropen Effekt.
 Die Anwendung von Digitalisglykosiden gegen Herzversagen wurde erstmals 1785 vom britischen Arzt William Withering beschrieben. Heilende Wirkungen der Herzglykoside sind allerdings schon seit dem Altertum bekannt.
Die Anwendung von Digitalisglykosiden gegen Herzversagen wurde erstmals 1785 vom britischen Arzt William Withering beschrieben. Heilende Wirkungen der Herzglykoside sind allerdings schon seit dem Altertum bekannt.


 Aktionspotentiale breiten sich über Kardiomyozyten und transversale Tubuli aus. Diaden / Triaden sind Kontaktstellen mit Ausläufern des endoplasmatischen Retikulums. Ca++ gelangt über L-Typ-Calciumkanäle (Dihydropyridinrezeptoren) in die Zelle, und über Ryanodinrezeptoren aus dem sarkoplasmatischen Retikulum (calciuminduzierte Calciumfreisetzung), das löst Kontraktion aus (elektromechanische Koppelung). Ein kleiner Teil des Calciums wird von Mitochondrien (30-35% des Zellvolumens) aufgenommen. In der Relaxationsphase wird Ca++
über die sarkoplasmatische Calciumpumpe (SERCA) in das Retikulum
zurück, und über Na/Ca-Austauscher nach extrazellulär befördert, Ca++ löst sich vom Troponin, die Kontraktion hört auf Aktionspotentiale breiten sich über Kardiomyozyten und transversale Tubuli aus. Diaden / Triaden sind Kontaktstellen mit Ausläufern des endoplasmatischen Retikulums. Ca++ gelangt über L-Typ-Calciumkanäle (Dihydropyridinrezeptoren) in die Zelle, und über Ryanodinrezeptoren aus dem sarkoplasmatischen Retikulum (calciuminduzierte Calciumfreisetzung), das löst Kontraktion aus (elektromechanische Koppelung). Ein kleiner Teil des Calciums wird von Mitochondrien (30-35% des Zellvolumens) aufgenommen. In der Relaxationsphase wird Ca++
über die sarkoplasmatische Calciumpumpe (SERCA) in das Retikulum
zurück, und über Na/Ca-Austauscher nach extrazellulär befördert, Ca++ löst sich vom Troponin, die Kontraktion hört auf
Ca++-Ionen
binden an Troponin C, Tropomyosin gibt die Reaktionsstellen für Myosinköpfe frei (Troponin T bindet an
Tropomyosin und unterstützt die Positionierung an das Aktin). Die Zahl aktivierter Querbrücken bestimmt die Kontraktionskraft des Herzmuskels. Die Ca++-Konzentration von 0,5-2 µM aktiviert nur einen Teil (im Ruhezustand ~40%) der
Myosin-Querbrücken; Steigerung des Calciumeinstroms (Adrenalin: ß1-Rezeptoren → [cAMP] → Proteinkinase A → Phosphorylierung spannungsabhängiger
Ca++-Kanäle → [Ca++]↑)
steigert die Schlagkraft (positive Inotropie). ~80% des in das
Sarkoplasma gelangten Ca++
stammt aus dem sarkoplasmatischen Retikulum, ~20% aus dem
Extrazellulärraum (dieses triggert die Freisetzung aus dem sarkoplasmatischen
Retikulum). Der Ca++-Sensor Triadin beeinflusst die Interaktion zwischen dem Ryanodinrezeptor und dem Ca++-bindenden Calsequestrin. Phospholamban bremst im dephosphorylierten Zustand die Ca++-Aufnahme in das Retikulum; seine Phosphorylierung hebt diese Bremswirkung auf und Ca++
wird in das Retikulum zurückgepumpt, insbesondere unter der Wirkung von
Katecholaminen, diese beschleunigen die Relaxation (Lusitropie)
Titin bestimmt die Dehnbarkeit der Sarkomere / des Myokards:
Myokardzellen werden nicht über 2,3 µm lang (mutiertes "Riesentitin" ermöglicht eine Dehnung der Sarkomere bis ~4 µm). Bei einem typischen enddiastolischen Volumen sind die Sarkomere 1,8-2,0 µm lang, die
höchste Kontraktionskraft erfolgt bei ~2,2 µm (starke Vordehnung):
Das Myokard arbeitet im ansteigenden Teil der Kraft-Volumen-Kurve, die
Kraftausbeute nimmt mit der diastolischen Füllung zu
Die Ruhedehnungskurve spiegelt mechanische Eigenschaften des Ventrikels wider. Eine
Kontraktion, die aus einer
iso(volu)metrischen und dann einer auxotonen Phase
besteht, bezeichnet man als Unterstützungszuckung (normaler Herzschlag). Deren Maximalwert
ist im Druck-Volumen-Diagramm durch die Lage auf der Kurve der U-Maxima
gekennzeichnet (jede Systole ist ein Maximum im Sinne, dass alle Muskelfasern an der Kontraktion teilnehmen). Die Distanz eines U-Maximums zum Ausgangspunkt auf der Ruhedehnungskurve gibt die Intensität der Kontraktionen an (Abschätzung der Inotropie)
Die
Geschwindigkeit der myokardialen Kontraktion hängt von der Belastung ab. Mit zunehmender Gegenkraft nimmt die
Verkürzungsgeschwindigkeit ab, was die Entleerung des betreffenden
Herzraumes verlangsamt, mit zunehmender Nachlast sinkt die
Auswurfgeschwindigkeit. Erhöhte Belastung der Ventrikel durch
wiederholtes intensives körperliches Training führt zu einem
"Sportlerherz", das Myokard nimmt von von ~300 g bis auf ~500 g zu;
durch erhöhten parasympathischen Einfluss kommt es zu
Ruhe-Bradykardie bis <40 bpm
Herzglykoside reduzieren die Aktivität der Na/K-Pumpe bis um 25%, intrazelluläres [Na+] steigt an, der treibende Natriumgradient für den Na/Ca-Austauscher sinkt, es wird weniger Ca++ exportiert (positiv inotroper Effekt). Der Rücktransport von Ca++ in das sarkoplasmatische Retikulum bleibt gleich (kein lusitroper Effekt)
|
 Die Informationen in dieser Website basieren auf verschiedenen Quellen:
Lehrbüchern, Reviews, Originalarbeiten u.a. Sie
sollen zur Auseinandersetzung mit physiologischen Fragen, Problemen und
Erkenntnissen anregen. Soferne Referenzbereiche angegeben sind, dienen diese zur Orientierung; die Grenzen sind aus biologischen, messmethodischen und statistischen Gründen nicht absolut. Wissenschaft fragt, vermutet und interpretiert; sie ist offen, dynamisch und evolutiv. Sie strebt nach Erkenntnis, erhebt aber nicht den Anspruch, im Besitz der "Wahrheit" zu sein.
Die Informationen in dieser Website basieren auf verschiedenen Quellen:
Lehrbüchern, Reviews, Originalarbeiten u.a. Sie
sollen zur Auseinandersetzung mit physiologischen Fragen, Problemen und
Erkenntnissen anregen. Soferne Referenzbereiche angegeben sind, dienen diese zur Orientierung; die Grenzen sind aus biologischen, messmethodischen und statistischen Gründen nicht absolut. Wissenschaft fragt, vermutet und interpretiert; sie ist offen, dynamisch und evolutiv. Sie strebt nach Erkenntnis, erhebt aber nicht den Anspruch, im Besitz der "Wahrheit" zu sein.



 Elektromechanische
Kopplung und Kontraktionsmechanismus
Elektromechanische
Kopplung und Kontraktionsmechanismus
 Connexin: nexus = Verknüpfung (nectere = fesseln, verbinden)
Connexin: nexus = Verknüpfung (nectere = fesseln, verbinden)
