

Eine Reise durch die Physiologie - Wie der Körper des Menschen funktioniert


 Transport,
Metabolismus und Clearance
Transport,
Metabolismus und Clearance
 Dynamik: δύναμις = Kraft
Dynamik: δύναμις = Kraft| Die
Konzentration eines Hormons / Wirkstoffs im Blut hängt von mehreren Faktoren ab: Anfangsmenge und
Produktionsrate, Aufnahme und Verteilung im Körper, Abbau und Ausscheidung (Clearance). Kinetik beschreibt, wie der Organismus
mit einem Stoff (Hormon, Medikament,..) verfährt. Unter Bioverfügbarkeit versteht man den Anteil des Hormons (Wirkstoffs), der ein Kompartiment (Kreislauf, Gewebe) erreicht, in dem es aktiv wird. Die biologische Halbwertszeit sagt aus, wie rasch ein in das System eingebrachter Stoff auf die Hälfte seiner Ausgangskonzentration abnimmt (durch Aufnahme in Zellen, Diffusion in die Blutbahn, Speicherung im Fettgewebe, Modifikation, Abbau, Ausscheidung). Die Dynamik eines Hormons beschreibt die Art und Weise seiner Wirkung auf den Organismus. Die Wirkung wird vermittelt durch Rezeptoraktivierung und zelluläre Folgevorgänge (wie Ioneneinstrom, Enzymaktivierung, Transkription, Synthesevorgänge). Ob das Hormon fett- (hydrophob) oder wasserlöslich (hydrophil) ist, spielt eine große Rolle: So werden hydrophile Hormone (z.B. Proteohormone) in Vesikeln gespeichert und können aus diesen bei Bedarf wieder freigesetzt werden; für lipophile (z.B. Steroide) kommt das nicht in Frage (Vesikelwände wären für sie kein Hindernis), sie werden durch Enzyminduktion neu synthetisiert. |
 (Pharmako)-Kinetik (Pharmako-) Dynamik
Applikationsformen
(Pharmako)-Kinetik (Pharmako-) Dynamik
Applikationsformen Kompartiment
Kompartiment 
 Core messages
Core messages Abbildung), und wenn sie
rasch abgebaut werden, bleiben Änderungen ihrer Konzentration im
Blutkreislauf u.U. unter der Nachweisbarkeitsgrenze. Bei endokriner
Funktionsweise ist der potentielle Adressat der hormonproduzierenden
Zellen der ganze Organismus. Das bedeutet aber nicht, dass die aktuelle
Konzentration eines Hormons ein direkter Indikator der Aktivität der
Hormondrüse ist; der Blutspiegel ist auch beeinflusst durch die
betreffende Kinetik, also Intensität und Zeitverlauf von Verteilung, Um- und Abbau sowie Entfernung des Hormons aus dem Körper. Abbildung: Informationsübertragung von Zelle zu Zelle
Abbildung), und wenn sie
rasch abgebaut werden, bleiben Änderungen ihrer Konzentration im
Blutkreislauf u.U. unter der Nachweisbarkeitsgrenze. Bei endokriner
Funktionsweise ist der potentielle Adressat der hormonproduzierenden
Zellen der ganze Organismus. Das bedeutet aber nicht, dass die aktuelle
Konzentration eines Hormons ein direkter Indikator der Aktivität der
Hormondrüse ist; der Blutspiegel ist auch beeinflusst durch die
betreffende Kinetik, also Intensität und Zeitverlauf von Verteilung, Um- und Abbau sowie Entfernung des Hormons aus dem Körper. Abbildung: Informationsübertragung von Zelle zu Zelle
 Zum Zeitpunkt der Messung anfänglich im Blut vorhandene Menge.
Wird ein Hormon in die Blutbahn abgegeben, verteilt es sich primär im
Plasma (beim Erwachsenen ~3 Liter).
Zum Zeitpunkt der Messung anfänglich im Blut vorhandene Menge.
Wird ein Hormon in die Blutbahn abgegeben, verteilt es sich primär im
Plasma (beim Erwachsenen ~3 Liter).  direkt zur
Verfügung. Produktionsrate: Durch Sekretion im hormonproduzierenden Gewebe hinzukommende Menge
direkt zur
Verfügung. Produktionsrate: Durch Sekretion im hormonproduzierenden Gewebe hinzukommende Menge  Einige Hormone werden kontinuierlich in den Extrazellulärraum abgegeben
(ohne spezifische Reizung: "basale Sekretionsrate"), die Produktion verstärkt sich auf
entsprechende Reize hin (z.B. Insulinanstieg infolge
Blutzuckerbelastung)
Andere (z.B. des hypothalamisch-hypophysären
Systems) gelangen pulsatil ins Blut (z.B. CRH, Somatoliberin). Hier ist es notwendig, Zeitprofile
der Hormonkonzentration im Blutplasma zu ermitteln, isolierte
Bestimmungen sind zufallsabhängig und nur sehr begrenzt aussagekräftig Größe
des Verteilungsvolumens (Konzentration = Menge / Volumen). Man unterscheidet verschiedene Verteilungsräume, weil sich Stoffe in diesen
Kompartimenten
unterschiedlich verteilen und verschieden schnell wieder aus
ihnen verschwinden. Zu berücksichtigen ist weiters, dass Stoffe, die sich in einem
Kompartiment verteilen, hier gebunden werden können (z.B. an
Transportproteine); nur ein Teil der Moleküle ist dann frei beweglich
und damit zwischen den Kompartimenten unmittelbar austauschbar
(
Einige Hormone werden kontinuierlich in den Extrazellulärraum abgegeben
(ohne spezifische Reizung: "basale Sekretionsrate"), die Produktion verstärkt sich auf
entsprechende Reize hin (z.B. Insulinanstieg infolge
Blutzuckerbelastung)
Andere (z.B. des hypothalamisch-hypophysären
Systems) gelangen pulsatil ins Blut (z.B. CRH, Somatoliberin). Hier ist es notwendig, Zeitprofile
der Hormonkonzentration im Blutplasma zu ermitteln, isolierte
Bestimmungen sind zufallsabhängig und nur sehr begrenzt aussagekräftig Größe
des Verteilungsvolumens (Konzentration = Menge / Volumen). Man unterscheidet verschiedene Verteilungsräume, weil sich Stoffe in diesen
Kompartimenten
unterschiedlich verteilen und verschieden schnell wieder aus
ihnen verschwinden. Zu berücksichtigen ist weiters, dass Stoffe, die sich in einem
Kompartiment verteilen, hier gebunden werden können (z.B. an
Transportproteine); nur ein Teil der Moleküle ist dann frei beweglich
und damit zwischen den Kompartimenten unmittelbar austauschbar
( Abbildung).
Abbildung).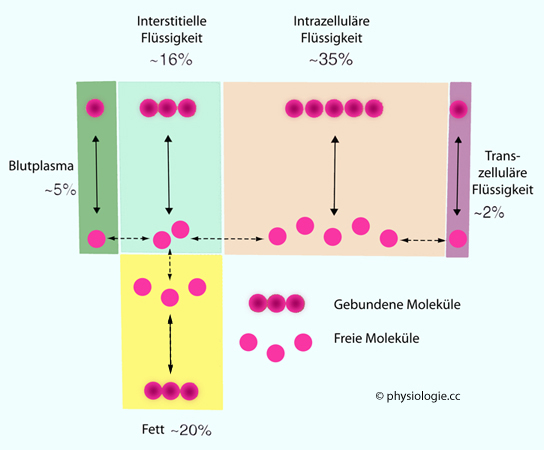 Abbildung: Flüssigkeitskompartimente im menschlichen Körper Als Kompartiment (fluid compartment)
im physiologischen / pharmakokinetischen Sinne bezeichnet man
Verteilungsräume für (von innen oder außen) eingebrachte Stoffe - Hormone, andere Stoffwechselprodukte, Pharmaka etc (über
Applikationsmöglichkeiten s. unten). Die wichtigsten sind - außer Blutplasma (~5% des Körpergewichts) -
das Interstitium (~16%), Fettgewebe (~20% - hier reichern sich vor allem lipophile Stoffe an, z.B. Steroidhormone),
intrazelluläre Flüssigkeit (~35%) und
transzelluläre Flüssigkeiten (~2,5% des Körpergewichts - third space:
Flüssigkeit in eigenen Räumen, wie z.B. Plauraspalt, Perikardialraum,
liquor cerebrospinalis, Kammerwasser,
Flüssigkeiten im Darm). Zwischen transzellulären Räumen und dem
Blutplasma steht immer eine Lage von Zellen (z.B. Pleuraepithel,
Ependymzellen im Gehirn, Kapillarendothel im Ziliarkörper), also eine
Schichte intrazellulärer Flüssigkeit.
Abbildung: Flüssigkeitskompartimente im menschlichen Körper Als Kompartiment (fluid compartment)
im physiologischen / pharmakokinetischen Sinne bezeichnet man
Verteilungsräume für (von innen oder außen) eingebrachte Stoffe - Hormone, andere Stoffwechselprodukte, Pharmaka etc (über
Applikationsmöglichkeiten s. unten). Die wichtigsten sind - außer Blutplasma (~5% des Körpergewichts) -
das Interstitium (~16%), Fettgewebe (~20% - hier reichern sich vor allem lipophile Stoffe an, z.B. Steroidhormone),
intrazelluläre Flüssigkeit (~35%) und
transzelluläre Flüssigkeiten (~2,5% des Körpergewichts - third space:
Flüssigkeit in eigenen Räumen, wie z.B. Plauraspalt, Perikardialraum,
liquor cerebrospinalis, Kammerwasser,
Flüssigkeiten im Darm). Zwischen transzellulären Räumen und dem
Blutplasma steht immer eine Lage von Zellen (z.B. Pleuraepithel,
Ependymzellen im Gehirn, Kapillarendothel im Ziliarkörper), also eine
Schichte intrazellulärer Flüssigkeit. Entscheidend für die Wirkung eines Hormons (Signalstoffs) ist seine Konzentration am Wirkungsort,
d.h. an den betreffenden Zellen. Dort läßt sich diese aber (im
Allgemeinen) nicht messen; man ist auf Konzentrationsbestimmungen in
Blut (oder anderen Körpersäften, z.B. Harn) angewiesen. Zur Frage, wie
die Substanz auf die Zellen und Gewebe einwirkt (Dynamik),
kommt das Problem, wie der Stoff sich im Körper verteilt und seine
Konzentration mit physiologischen Vorgängen (Synthese, Verteilung,
Aktivierung / Inaktivierung, Abbau, Ausscheidung) zu- und wieder
abnimmt (Kinetik). (Pharmakokinetik) beschreibt die Vorgänge, denen ein Wirkstoff (Hormon, Medikament ..)
bei seiner Reise durch den Organismus unterliegt. Sie umfasst Absorption, Distribution, Metabolisierung und Clearance:
Entscheidend für die Wirkung eines Hormons (Signalstoffs) ist seine Konzentration am Wirkungsort,
d.h. an den betreffenden Zellen. Dort läßt sich diese aber (im
Allgemeinen) nicht messen; man ist auf Konzentrationsbestimmungen in
Blut (oder anderen Körpersäften, z.B. Harn) angewiesen. Zur Frage, wie
die Substanz auf die Zellen und Gewebe einwirkt (Dynamik),
kommt das Problem, wie der Stoff sich im Körper verteilt und seine
Konzentration mit physiologischen Vorgängen (Synthese, Verteilung,
Aktivierung / Inaktivierung, Abbau, Ausscheidung) zu- und wieder
abnimmt (Kinetik). (Pharmakokinetik) beschreibt die Vorgänge, denen ein Wirkstoff (Hormon, Medikament ..)
bei seiner Reise durch den Organismus unterliegt. Sie umfasst Absorption, Distribution, Metabolisierung und Clearance: Niere (Harn): Glomeruläre Filtration ungebundener Moleküle, Sekretion in proximalen Tubuli. Über distale Tubuli kann Rückresorption in die Blutbahn erfolgenDarm (Stuhl), Leber (Galle), Haut (Schweiß), Atmung (Exspirationsluft), vor allem pulmonal applizierter Substanzen (Inhalation),Speichel, TränenMuttermilch (kann unerwünschte Effekte beim Baby bewirken)
Niere (Harn): Glomeruläre Filtration ungebundener Moleküle, Sekretion in proximalen Tubuli. Über distale Tubuli kann Rückresorption in die Blutbahn erfolgenDarm (Stuhl), Leber (Galle), Haut (Schweiß), Atmung (Exspirationsluft), vor allem pulmonal applizierter Substanzen (Inhalation),Speichel, TränenMuttermilch (kann unerwünschte Effekte beim Baby bewirken)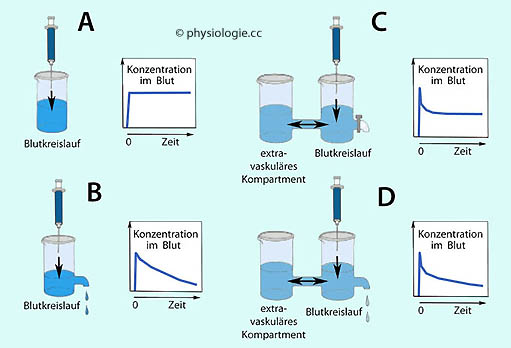 Abbildung: Einfache Modelle der Verteilungskinetik Abbildung), wie der
anschließende Zeitverlauf der Stoffkonzentration im Blut aussehen muss:
Verbleibt der Stoff in der Blutbahn (keine transvasale Durchgängigkeit)
und wird er auch nicht aus dem Blut entfernt (kein Abbau), steigt seine
Konzentration auf einen gleichbleibenden Endwert (A; hypothetisch)
Wird er aus dem Blut entfernt, ohne in andere Kompartimente zu
gelangen, nimmt die Konzentration nach Erreichen eines Gipfelwertes
stetig ab (B, ebenfalls hypothetisch)
Verteilt er sich vom Blutkreislauf her in einem "zweiten" extravasalen
Kompartiment (im Gewebe), ohne aber abgebaut oder ausgeschieden zu werden, nimmt seine Konzentration nach der
Verteilungszeit nicht weiter ab (C; auch dieser Fall ist hypothetisch) Verteilt er sich z.T. außerhalb der Blutbahn im Körper (hier als ein
Kompartiment angenommen, d.h. der gesamte Organismus hätte
eine für alle Organe gültige Distributionscharakteristik) und wird
gleichzeitig abgebaut / ausgeschieden, ergibt sich der in D gezeigte ZeitverlaufAbbildung).
Abbildung: Einfache Modelle der Verteilungskinetik Abbildung), wie der
anschließende Zeitverlauf der Stoffkonzentration im Blut aussehen muss:
Verbleibt der Stoff in der Blutbahn (keine transvasale Durchgängigkeit)
und wird er auch nicht aus dem Blut entfernt (kein Abbau), steigt seine
Konzentration auf einen gleichbleibenden Endwert (A; hypothetisch)
Wird er aus dem Blut entfernt, ohne in andere Kompartimente zu
gelangen, nimmt die Konzentration nach Erreichen eines Gipfelwertes
stetig ab (B, ebenfalls hypothetisch)
Verteilt er sich vom Blutkreislauf her in einem "zweiten" extravasalen
Kompartiment (im Gewebe), ohne aber abgebaut oder ausgeschieden zu werden, nimmt seine Konzentration nach der
Verteilungszeit nicht weiter ab (C; auch dieser Fall ist hypothetisch) Verteilt er sich z.T. außerhalb der Blutbahn im Körper (hier als ein
Kompartiment angenommen, d.h. der gesamte Organismus hätte
eine für alle Organe gültige Distributionscharakteristik) und wird
gleichzeitig abgebaut / ausgeschieden, ergibt sich der in D gezeigte ZeitverlaufAbbildung). Abbildung: Modell zur mathematischen Beschreibung der Verteilungskinetik zwischen mehreren Körperkompartimenten
Abbildung: Modell zur mathematischen Beschreibung der Verteilungskinetik zwischen mehreren Körperkompartimenten Über Kinetik und Modellierung von Stoffverteilungen im Körper s. auch dort Verteilungsvolumen (distribution volume): In welchen Räumen (Kompartimenten, Compartments)
löst sich das Hormon / die Wirksubstanz in welcher Zeit? Beispielsweise
würde eine vollständige (und gleichmäßige) Verteilung im
Gesamt-Körperwasser (TBW: total body water) ein Verteilungsvolumen bedeuten, das ~60% des
Körpergewichts entspricht. Unter Bioverfügbarkeit (bioavailability) versteht man den Anteil des Hormons / Wirkstoffs, der den systemischen Kreislauf
und damit das Gewebe erreicht, an dem es aktiv werden kann. Sobald das
Hormon in das Blut gelangt, ist seine Bioverfügbarkeit nach dieser
Definition 100%. Ein im Fettgewebe gespeicherter Anteil (z.B. ein
Steroid) wäre hingegen zwar im Körper vorhanden, aber zum Zeitpunkt
seiner Speicherung nicht bioverfügbar. (Bei Arzneistoffen stellt sich
die Frage der Verabreichung: Wie rasch wird das Pharmakon resorbiert?
Wie stark ist der first-pass-Effekt durch die Leber?) Clearance:
Das ist die Plasmamenge, aus der die betreffende Substanz in einer bestimmten Zeit (rechnerisch)
vollständig verschwunden ist, d.h. das Plasma wurde von ihr in der
betreffenden Zeit "gereinigt" (cleared). Die Niere ist ein Hauptorgan der "Reinigung" (Ausscheidung) von Stoffen, und man unterscheidet daher eine renale (ClR) von einer nicht-renalen (insbesondere hepatischen) Clearance (ClNR). Zur renalen Clearance s. dort. Abbau (z.B. Leber) und Ausscheidung (Niere) bestimmen die biologische Halbwertszeit,
d.h. die (virtuelle) Dauer ab einem völligen Aufhören der Nachlieferung
eines Hormons bis zum Erreichen der halben Serumkonzentration
(nichtlinearer Zeitverlauf beginnend mit 100% = Anfangskonzentration,
z.B. 50% nach Ablauf einer Halbwertszeit, 25% nach Ablauf der doppelten
Zeit usw.). Proteohormone werden vorwiegend von Proteasen im Blut und von Tubuluszellen in der Niere (nach Filtration und Rückresorption) abgebaut, Steroide werden in der Leber konjugiert (Biotransformation: Glukuronierung, Sulfatierung) und dann über Niere und Darm ausgeschieden.
Über Kinetik und Modellierung von Stoffverteilungen im Körper s. auch dort Verteilungsvolumen (distribution volume): In welchen Räumen (Kompartimenten, Compartments)
löst sich das Hormon / die Wirksubstanz in welcher Zeit? Beispielsweise
würde eine vollständige (und gleichmäßige) Verteilung im
Gesamt-Körperwasser (TBW: total body water) ein Verteilungsvolumen bedeuten, das ~60% des
Körpergewichts entspricht. Unter Bioverfügbarkeit (bioavailability) versteht man den Anteil des Hormons / Wirkstoffs, der den systemischen Kreislauf
und damit das Gewebe erreicht, an dem es aktiv werden kann. Sobald das
Hormon in das Blut gelangt, ist seine Bioverfügbarkeit nach dieser
Definition 100%. Ein im Fettgewebe gespeicherter Anteil (z.B. ein
Steroid) wäre hingegen zwar im Körper vorhanden, aber zum Zeitpunkt
seiner Speicherung nicht bioverfügbar. (Bei Arzneistoffen stellt sich
die Frage der Verabreichung: Wie rasch wird das Pharmakon resorbiert?
Wie stark ist der first-pass-Effekt durch die Leber?) Clearance:
Das ist die Plasmamenge, aus der die betreffende Substanz in einer bestimmten Zeit (rechnerisch)
vollständig verschwunden ist, d.h. das Plasma wurde von ihr in der
betreffenden Zeit "gereinigt" (cleared). Die Niere ist ein Hauptorgan der "Reinigung" (Ausscheidung) von Stoffen, und man unterscheidet daher eine renale (ClR) von einer nicht-renalen (insbesondere hepatischen) Clearance (ClNR). Zur renalen Clearance s. dort. Abbau (z.B. Leber) und Ausscheidung (Niere) bestimmen die biologische Halbwertszeit,
d.h. die (virtuelle) Dauer ab einem völligen Aufhören der Nachlieferung
eines Hormons bis zum Erreichen der halben Serumkonzentration
(nichtlinearer Zeitverlauf beginnend mit 100% = Anfangskonzentration,
z.B. 50% nach Ablauf einer Halbwertszeit, 25% nach Ablauf der doppelten
Zeit usw.). Proteohormone werden vorwiegend von Proteasen im Blut und von Tubuluszellen in der Niere (nach Filtration und Rückresorption) abgebaut, Steroide werden in der Leber konjugiert (Biotransformation: Glukuronierung, Sulfatierung) und dann über Niere und Darm ausgeschieden.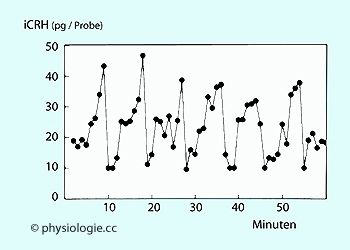 Abbildung: Zeitverlauf von Kortikoliberin im Blut Abbildung: Zeitverlauf CRH imBlut). Wird
der Stoff glomerulär filtriert? Wenn nicht (weil er proteingebunden ist
- z.B. Schilddrüsenhormone - oder weil das Molekül wegen seiner Größe
(oder Ladung) die Kapillarwand kaum durchdringt - z.B. Proteohormone),
gelangt auch kaum etwas in Tubulussystem und Harn Was geschieht
mit dem Stoff entlang des Tubulussystems? Interessieren sich die
Tubuluszellen nicht für ihn, weil sie über kein passendes
Transportsystem verfügen, dann wird er weder rückresorbiert noch
sezerniert (Beispiel Inulin), und automatisch wird er im Harn etwa
100-fach konzentriert (da Wasser zu 99% aus den Tubuli zurückgewonnen
wird, s. dort) Gibt es eine Rückresorption (gilt für fast alle filtrierten Stoffe,
auch Hormone), dann wird die eben erwähnte Anreicherung im Harn nur
schwächer oder gar nicht stattfinden Umgekehrt kann es sein, dass der Stoff sezerniert wird (z.B. in den distalen Tubuli), wie das für zahlreiche Medikamente zutrifft; das steigert wiederum die Konzentration des Stoffes im Harn. im Körper. Dabei interessieren verschiedene Fragen, wie Welche Rezeptoren werden aktiviert?
Welcher Mechanismus spielt die Hauptrolle (Membrankanäle, Enzyme,
Gen(in)aktivierung)? In welcher Weise ist die biologische Wirkung von Gewebe, Dosis
und Zeitverlauf abhängig?
Hormone entfalten ihre Wirkung, indem sie sich an (membranständige oder zytoplasmatische) Rezeptormoleküle anlagern. Binden sie an
der Zellmembran, werden sie typischerweise mit dem
Rezeptor endozytiert (reduzierter Rezeptorbesatz an der Membran, dadurch verringerte Hormonempfindlichkeit der Zelle: receptor downregulation),
dissoziiert dann vom Rezeptor und wird an die Außenmembran rückgeführt (receptor upregulation), der Signalstoff endo- und lysosomal abgebaut. Hormone,
die an Rezeptor- oder
Transportmoleküle in der Zellmembran gebunden werden, können z.T.
intrazellulär vesikulär gespeichert und bei Bedarf rasch freigesetzt
werden. Mobilisierung dieser Reserve erfolgt umgehend
(Peptidhormone).
Anders bei
lipophilen Signalstoffen: Diese werden nicht in Vesikeln
gespeichert,
sondern bei Bedarf frisch synthetisiert. Die Hormonproduktion ist hier
der begrenzende Faktor, die Freisetzung erfolgt verzögert (Steroidhormone).
Abbildung: Zeitverlauf von Kortikoliberin im Blut Abbildung: Zeitverlauf CRH imBlut). Wird
der Stoff glomerulär filtriert? Wenn nicht (weil er proteingebunden ist
- z.B. Schilddrüsenhormone - oder weil das Molekül wegen seiner Größe
(oder Ladung) die Kapillarwand kaum durchdringt - z.B. Proteohormone),
gelangt auch kaum etwas in Tubulussystem und Harn Was geschieht
mit dem Stoff entlang des Tubulussystems? Interessieren sich die
Tubuluszellen nicht für ihn, weil sie über kein passendes
Transportsystem verfügen, dann wird er weder rückresorbiert noch
sezerniert (Beispiel Inulin), und automatisch wird er im Harn etwa
100-fach konzentriert (da Wasser zu 99% aus den Tubuli zurückgewonnen
wird, s. dort) Gibt es eine Rückresorption (gilt für fast alle filtrierten Stoffe,
auch Hormone), dann wird die eben erwähnte Anreicherung im Harn nur
schwächer oder gar nicht stattfinden Umgekehrt kann es sein, dass der Stoff sezerniert wird (z.B. in den distalen Tubuli), wie das für zahlreiche Medikamente zutrifft; das steigert wiederum die Konzentration des Stoffes im Harn. im Körper. Dabei interessieren verschiedene Fragen, wie Welche Rezeptoren werden aktiviert?
Welcher Mechanismus spielt die Hauptrolle (Membrankanäle, Enzyme,
Gen(in)aktivierung)? In welcher Weise ist die biologische Wirkung von Gewebe, Dosis
und Zeitverlauf abhängig?
Hormone entfalten ihre Wirkung, indem sie sich an (membranständige oder zytoplasmatische) Rezeptormoleküle anlagern. Binden sie an
der Zellmembran, werden sie typischerweise mit dem
Rezeptor endozytiert (reduzierter Rezeptorbesatz an der Membran, dadurch verringerte Hormonempfindlichkeit der Zelle: receptor downregulation),
dissoziiert dann vom Rezeptor und wird an die Außenmembran rückgeführt (receptor upregulation), der Signalstoff endo- und lysosomal abgebaut. Hormone,
die an Rezeptor- oder
Transportmoleküle in der Zellmembran gebunden werden, können z.T.
intrazellulär vesikulär gespeichert und bei Bedarf rasch freigesetzt
werden. Mobilisierung dieser Reserve erfolgt umgehend
(Peptidhormone).
Anders bei
lipophilen Signalstoffen: Diese werden nicht in Vesikeln
gespeichert,
sondern bei Bedarf frisch synthetisiert. Die Hormonproduktion ist hier
der begrenzende Faktor, die Freisetzung erfolgt verzögert (Steroidhormone). Ein Teil wird inaktiviert (enzymatischer Abbau) oder von Zellen aufgenommen (wenn es die sezernierenden Zellen sind, spricht man von reuptake) Ein Teil diffundiert vom Ort der
Freisetzung weg, gelangt in den Kreislauf und ist dann im Blut
nachweisbar (spillover) - z.B. Hormonwerte, an denen sich der Kliniker (zwangsläufig) orientiert Viele Hormone werden im weiteren Verlauf modifiziert (z.B. in der Leber: Biotransformation) Letztlich
werden sie (oder ihre Abbauprodukte) aus dem Körper entfernt (Harn,
Galle / Stuhl, Schweiss, andere Sekrete, Atemluft, Blut, Hautschuppen,
Haare) Peptidhormone
werden durch Proteolyse abgebaut (z.B. in Leber oder Niere),
Katecholamine durch Monoaminooxidase (MAO) und
Catechol-O-Methyltransferase (COMT), Steroidhormone durch
Biotransformation in wasserlöslichere Metabolite umgewandelt und diese
mit Harn und Galle ausgeschieden. Schilddrüsenhormone werden dejodiniert und durchlaufen in der Leber
mehrere Biotransformationen
(wie Decarboxylierung, Glukuronierung). Sie sind zu einem hohen Prozentsatz an
Plasmaeiweiß gebunden und haben lange Halbwertszeiten (mehrere Tage).
Ein Teil wird inaktiviert (enzymatischer Abbau) oder von Zellen aufgenommen (wenn es die sezernierenden Zellen sind, spricht man von reuptake) Ein Teil diffundiert vom Ort der
Freisetzung weg, gelangt in den Kreislauf und ist dann im Blut
nachweisbar (spillover) - z.B. Hormonwerte, an denen sich der Kliniker (zwangsläufig) orientiert Viele Hormone werden im weiteren Verlauf modifiziert (z.B. in der Leber: Biotransformation) Letztlich
werden sie (oder ihre Abbauprodukte) aus dem Körper entfernt (Harn,
Galle / Stuhl, Schweiss, andere Sekrete, Atemluft, Blut, Hautschuppen,
Haare) Peptidhormone
werden durch Proteolyse abgebaut (z.B. in Leber oder Niere),
Katecholamine durch Monoaminooxidase (MAO) und
Catechol-O-Methyltransferase (COMT), Steroidhormone durch
Biotransformation in wasserlöslichere Metabolite umgewandelt und diese
mit Harn und Galle ausgeschieden. Schilddrüsenhormone werden dejodiniert und durchlaufen in der Leber
mehrere Biotransformationen
(wie Decarboxylierung, Glukuronierung). Sie sind zu einem hohen Prozentsatz an
Plasmaeiweiß gebunden und haben lange Halbwertszeiten (mehrere Tage). Abbildung). Sie können auf die Zone der Aufbringung begrenzt bleiben und hier wirken, sich über limitierte Strecken
verteilen (via Interstitium, Liquor cerebrospinalis) oder
im Körper verteilen (über den
Kreislauf). Ihre Resorption kann von vielen Faktoren abhängen, wie
Beschaffenheit des zu
resorbierendern Stoffes (Partikelgröße, Löslichkeit etc),
Begleitstoffen, Darmmotilität, verfügbaren Transportmechanismen. Die
Verteilung hängt von physiologischen Eigenschaften der (z.T.
virtuellen) Kompartimente ab, in denen sie stattfindet.
Abbildung). Sie können auf die Zone der Aufbringung begrenzt bleiben und hier wirken, sich über limitierte Strecken
verteilen (via Interstitium, Liquor cerebrospinalis) oder
im Körper verteilen (über den
Kreislauf). Ihre Resorption kann von vielen Faktoren abhängen, wie
Beschaffenheit des zu
resorbierendern Stoffes (Partikelgröße, Löslichkeit etc),
Begleitstoffen, Darmmotilität, verfügbaren Transportmechanismen. Die
Verteilung hängt von physiologischen Eigenschaften der (z.T.
virtuellen) Kompartimente ab, in denen sie stattfindet. Abbildung: Wege der Applikation von Medikamenten Über die Bindehaut des Auges (intraokular; Augentropfen) Inhalation: Pulmonal (Zufuhr über die Atemwege, hauptsächlich topische Wirkung - z.B. bei Infektionen der Atemwege) / nasal (Nasenspray, Resorption erfolgt
wahrscheinlich auch über lymphatisches Gewebe der Nasenschleimhaut - z.B. Vasopressin, Calcitonin, GnRH).
Über die Schleimhaut von Zunge (sublingual) und Mundhöhle (oromukös) bzw. über die Wangenschleimhaut (buccal). Die Schleimhaut dieser Region ist sehr gut durchblutet, Stoffe können rasch resorbiert werden. Blut aus
diesem Bereich gelangt in die obere Hohlvene, also direkt in den
Kreislauf (nicht über Pfortaderkreislauf und Leber, kein First-pass-Effekt) Über Magen und Dünndarm (orale Einnahme von Kapseln / Tabletten, enterale Resorption). Dabei sind die Stoffe den Verdauungsvorgängen ausgesetzt; sie können noch vor ihrer Absorption
durch die Darmmukosa abgebaut (z.B. Proteohormone) oder verändert
werden, entweder im Darm oder nach Passage durch die Pfortader in der
Leber (Biotransformation), was unter Umständen pharmakologische Aktivierung oder Inaktivierung zur Folge haben kann
Intravenös (i.v. - systemisch), wobei die Substanz verlässlich und rasch in die Blutbahn eingebracht wird
(Bolus, vorübergehend hohe Konzentration). Verwendung von Pumpen
verlangsamt und stabilisiert den Prozess Intramuskulär
(i.m. - Depot); Muskeln sind gut mit Gefäßen versorgt, injizierte
Substanzen werden rasch resorbiert und ihr Effekt tritt rascher auf als
bei oraler Gabe. Stoffe aus wässriger Lösung werden rasch, aus öliger
Suspension langsam resorbiert ("Depotpräparate")
Subkutan (s.c.): Bei diesen Applikationsformen hängt die
Ankunft im Kreislauf von der Diffusion durch das Gewebe sowie die
Stärke der Durchblutung ab.
Das Unterhautgewebe ist weniger gut mit Gefäßen versorgt als z.B. die
Muskulatur, die Resorption kann daher langsamer verlaufen
Transdermal (cutaneous
- Creme, Pflaster..). Die Substanz wird je nach Fettlöslichkeit unterschiedlich rasch resorbiert (Suspension in öliger Grundlage), gelangt in den Kreislauf und wirkt systemisch (z.B. Steroide, Ibuprofen) Rektal (Suppositorien) - Rektal resorbierte Substanzen gelangen (über die inneren Hämorrhoidalvenen) etwa zur Hälfte in den Pfortaderkreislauf und damit zur Leber (First-pass-Effekt),
etwa zur Hälfte daran vorbei (über die äußeren Hämorrhoidalvenen) direkt in den Kreislauf. Die Resorption
kann unvollständig erfolgen, die Dosierung ist dadurch beeinträchtigt
Andere, z.B. intraarteriell, aural (Gehörgang), epidural (Kanüle), intrathekal (in den Liquor des Subarachnoidalraums), intravitreal
(in den Glaskörper), intrakardial (Herzinjektion), intraartikulär (in
ein Gelenk), intraperitoneal (i.p. - Injektion, Infusion), vaginal. Inhalation: Oral oder nasal, Resorption über das respiratorische Epithel. Vorteile: Rasche Aufnahme und Wirkung, weniger systemische Nebenwirkungen. Nachteil: Anwendung muß geübt werden Intrathekale
Injektion: Über den Liquor. Vorteile: Rasch und effektiv; Pharmaka, welche die
Blut-Hirn-Schranke nicht überqueren können, gelangen direkt zum ZNS. Nachteile: Invasiv, erfordert medizinisches Personal mit spezieller Erfahrung Lokale Aufbringung auf Haut oder Schleimhaut (topisch). Vorteile: Nichtinvasiv, schmerzfrei, wenige Nebeneffekte. Nachteil: Anwendung auf Haut und oberflächliche Mukosa beschränkt Oral: Aufnahme über GI-System, gebräuchlichste Form der Verabreichung. Vorteile: Einfach, kostengünstig, schmerzlos, kann durch Magenspülung wieder entfernt werden. Nachteile:
Kooperation der Person erforderlich, schwierig bis unmöglich bei
Erbrechen oder Bewusstlosigkeit, Absorption hängt von mehreren Faktoren
ab (unsicher), langsamer Wirkungseintritt (nicht für Notfälle geeignet) Sublingual / buccal: Über Schleimhaut der Zunge bzw. der Backentaschen. Vorteile: Ähnlich wie oral, rasche Aufnahme und Wirkung, kein Abbau durch Verdauungsenzyme, kein first-pass-Effekt der Leber. Nachteile: Absorption unsicher, effektiv nur für spezifische Pharmake (z.B. lipophil) Rektal: Abtransport über Hämorrhoidalvenensystem. Vorteile:
Bei Bewusstlosen möglich, rasche Aufnahme, die Substanz entgeht
enzymatischen Einflüssen des Verdauungsystems, die Hälfte davon umgeht
den hepatischen first-pass. Nachteile: Unangenehm, Kooperation der Person meist erforderlich, Aufnahme der Substanz unsicher Subkutan: Einbringen unter die Haut. Vorteile: Vermeiden des First-pass-Effekts; langsamer, anhaltender, voraussagbarer Wirkungseintritt. Nachteile: Invasiv, auf kleine Mengen beschränkt Intravenös: Injektion (Bolus) oder Infusion. Vorteile:
Sofortiger Wirkungseintritt, präzise Dosierung, für Notfälle geeignet,
Umgehung metabolischer Abbauvorgänge (GI-Enzyme, first pass),
Einstellen gewünschter Plasmakonzentration möglich. Nachteile:
Invasiv, schmerzhaft, medizinisches Personal erforderlich, Infektions-
und Kontaminationsgefahr, evt. Thrombophlebitis, mögliche toxische
Wirkungen schwer behandelbar Intramuskulär: Injektion in Muskelgewebe. Vorteile:
Umgehung metabolischer Abbauvorgänge (GI-Enzyme, first pass), Absorptionsrate beeinflussbar. Nachteile:
Invasiv, schmerzhaft, medizinisches Personal erforderlich, kann
Muskalkrämpfe auslösen, kann Blutungen hervorrufen (bei
Antikoagulantieneinnahme nicht empfohlen) Transdermal: Als Pflaster, das den Wirkstoff über Stunden / Tage langsam an den Kreislauf abgibt. Vorteile:
Schmerzfrei, einfach, Umgehung des Gastrointestinaltrakts. Nachteile:
Kann Hautirritationen bewirken, nur für lipophile Substanzen in
niedriger Dosierung
Abbildung: Wege der Applikation von Medikamenten Über die Bindehaut des Auges (intraokular; Augentropfen) Inhalation: Pulmonal (Zufuhr über die Atemwege, hauptsächlich topische Wirkung - z.B. bei Infektionen der Atemwege) / nasal (Nasenspray, Resorption erfolgt
wahrscheinlich auch über lymphatisches Gewebe der Nasenschleimhaut - z.B. Vasopressin, Calcitonin, GnRH).
Über die Schleimhaut von Zunge (sublingual) und Mundhöhle (oromukös) bzw. über die Wangenschleimhaut (buccal). Die Schleimhaut dieser Region ist sehr gut durchblutet, Stoffe können rasch resorbiert werden. Blut aus
diesem Bereich gelangt in die obere Hohlvene, also direkt in den
Kreislauf (nicht über Pfortaderkreislauf und Leber, kein First-pass-Effekt) Über Magen und Dünndarm (orale Einnahme von Kapseln / Tabletten, enterale Resorption). Dabei sind die Stoffe den Verdauungsvorgängen ausgesetzt; sie können noch vor ihrer Absorption
durch die Darmmukosa abgebaut (z.B. Proteohormone) oder verändert
werden, entweder im Darm oder nach Passage durch die Pfortader in der
Leber (Biotransformation), was unter Umständen pharmakologische Aktivierung oder Inaktivierung zur Folge haben kann
Intravenös (i.v. - systemisch), wobei die Substanz verlässlich und rasch in die Blutbahn eingebracht wird
(Bolus, vorübergehend hohe Konzentration). Verwendung von Pumpen
verlangsamt und stabilisiert den Prozess Intramuskulär
(i.m. - Depot); Muskeln sind gut mit Gefäßen versorgt, injizierte
Substanzen werden rasch resorbiert und ihr Effekt tritt rascher auf als
bei oraler Gabe. Stoffe aus wässriger Lösung werden rasch, aus öliger
Suspension langsam resorbiert ("Depotpräparate")
Subkutan (s.c.): Bei diesen Applikationsformen hängt die
Ankunft im Kreislauf von der Diffusion durch das Gewebe sowie die
Stärke der Durchblutung ab.
Das Unterhautgewebe ist weniger gut mit Gefäßen versorgt als z.B. die
Muskulatur, die Resorption kann daher langsamer verlaufen
Transdermal (cutaneous
- Creme, Pflaster..). Die Substanz wird je nach Fettlöslichkeit unterschiedlich rasch resorbiert (Suspension in öliger Grundlage), gelangt in den Kreislauf und wirkt systemisch (z.B. Steroide, Ibuprofen) Rektal (Suppositorien) - Rektal resorbierte Substanzen gelangen (über die inneren Hämorrhoidalvenen) etwa zur Hälfte in den Pfortaderkreislauf und damit zur Leber (First-pass-Effekt),
etwa zur Hälfte daran vorbei (über die äußeren Hämorrhoidalvenen) direkt in den Kreislauf. Die Resorption
kann unvollständig erfolgen, die Dosierung ist dadurch beeinträchtigt
Andere, z.B. intraarteriell, aural (Gehörgang), epidural (Kanüle), intrathekal (in den Liquor des Subarachnoidalraums), intravitreal
(in den Glaskörper), intrakardial (Herzinjektion), intraartikulär (in
ein Gelenk), intraperitoneal (i.p. - Injektion, Infusion), vaginal. Inhalation: Oral oder nasal, Resorption über das respiratorische Epithel. Vorteile: Rasche Aufnahme und Wirkung, weniger systemische Nebenwirkungen. Nachteil: Anwendung muß geübt werden Intrathekale
Injektion: Über den Liquor. Vorteile: Rasch und effektiv; Pharmaka, welche die
Blut-Hirn-Schranke nicht überqueren können, gelangen direkt zum ZNS. Nachteile: Invasiv, erfordert medizinisches Personal mit spezieller Erfahrung Lokale Aufbringung auf Haut oder Schleimhaut (topisch). Vorteile: Nichtinvasiv, schmerzfrei, wenige Nebeneffekte. Nachteil: Anwendung auf Haut und oberflächliche Mukosa beschränkt Oral: Aufnahme über GI-System, gebräuchlichste Form der Verabreichung. Vorteile: Einfach, kostengünstig, schmerzlos, kann durch Magenspülung wieder entfernt werden. Nachteile:
Kooperation der Person erforderlich, schwierig bis unmöglich bei
Erbrechen oder Bewusstlosigkeit, Absorption hängt von mehreren Faktoren
ab (unsicher), langsamer Wirkungseintritt (nicht für Notfälle geeignet) Sublingual / buccal: Über Schleimhaut der Zunge bzw. der Backentaschen. Vorteile: Ähnlich wie oral, rasche Aufnahme und Wirkung, kein Abbau durch Verdauungsenzyme, kein first-pass-Effekt der Leber. Nachteile: Absorption unsicher, effektiv nur für spezifische Pharmake (z.B. lipophil) Rektal: Abtransport über Hämorrhoidalvenensystem. Vorteile:
Bei Bewusstlosen möglich, rasche Aufnahme, die Substanz entgeht
enzymatischen Einflüssen des Verdauungsystems, die Hälfte davon umgeht
den hepatischen first-pass. Nachteile: Unangenehm, Kooperation der Person meist erforderlich, Aufnahme der Substanz unsicher Subkutan: Einbringen unter die Haut. Vorteile: Vermeiden des First-pass-Effekts; langsamer, anhaltender, voraussagbarer Wirkungseintritt. Nachteile: Invasiv, auf kleine Mengen beschränkt Intravenös: Injektion (Bolus) oder Infusion. Vorteile:
Sofortiger Wirkungseintritt, präzise Dosierung, für Notfälle geeignet,
Umgehung metabolischer Abbauvorgänge (GI-Enzyme, first pass),
Einstellen gewünschter Plasmakonzentration möglich. Nachteile:
Invasiv, schmerzhaft, medizinisches Personal erforderlich, Infektions-
und Kontaminationsgefahr, evt. Thrombophlebitis, mögliche toxische
Wirkungen schwer behandelbar Intramuskulär: Injektion in Muskelgewebe. Vorteile:
Umgehung metabolischer Abbauvorgänge (GI-Enzyme, first pass), Absorptionsrate beeinflussbar. Nachteile:
Invasiv, schmerzhaft, medizinisches Personal erforderlich, kann
Muskalkrämpfe auslösen, kann Blutungen hervorrufen (bei
Antikoagulantieneinnahme nicht empfohlen) Transdermal: Als Pflaster, das den Wirkstoff über Stunden / Tage langsam an den Kreislauf abgibt. Vorteile:
Schmerzfrei, einfach, Umgehung des Gastrointestinaltrakts. Nachteile:
Kann Hautirritationen bewirken, nur für lipophile Substanzen in
niedriger Dosierung 
 Der Blutspiegel einer Substanz hängt ab von der im Kreislauf vorhandene Anfangsmenge, der durch Sekretion hinzukommenden und durch Speicherung, Abbau und Ausscheidung
verschwindenden Menge. Die Substanz kann mehr oder weniger intensiv an
Plasmaeiweiß gebunden sein; die gebundene Fraktion verbleibt
(weitgehend) im Kreislauf. Mit dem Anteil proteingebundenen Hormons
steigt seine Halbwertszeit. Die
Wirkung eines Signalstoffs hängt ab von seiner Konzentration am
Wirkungsort, der Ausstattung und Zugänglichkeit der Empfängerzellen mit
Rezeptoren, sowie den zwischen Rezeptor und Effekt geschalteten
Mechanismen Kinetik umfasst
Resorption, Transport, Distribution, Metabolisierung (Aktivierung /
Inaktivierung, Konjugation, Abbau) und Exkretion. Das Zeitprofil einer
Substanz nach ihrem Einbringen in den Kreislauf hängt von solchen
kinetischen Faktoren ab, z.B. der Permeabilität der Gefäßwände für
diesen Stoff. Die Verteilungskinetik lässt sich bei Kenntnis der wesentlichen Parameter - Bioverfügbarkeit, Verteilungsvolumen, Clearance, Halbwertszeit - mathematisch
modellieren. Komplexe Modelle berücksichtigen unterschiedliche
Eigenschaften verschiedener Organe und Gewebe. Die Art von
Proben, die für Konzentrationsmessungen herangezogen werden (Blut, Harn
etc), bestimmt auch die Art der Abbildung des Konzentrations- und Zeitverlaufs Dynamik
beschäftigt sich mit der Aktivität einer Wirksubstanz im Körper: Welche
Rezeptoren werden aktiviert? Welcher Mechanismus tritt anschließend in
Kraft (Membrankanäle, Enzyme, Gen(in)aktivierung)? Wie hängt die
biologische Wirkung von Gewebe, Dosis und Zeitverlauf ab? Peptidhormone werden enzymatisch abgebaut, Schilddrüsenhormone dejodiniert, Steroidhormone durch Biotransformation in wasserlöslichere Metabolite umgewandelt und diese mit Harn und Galle ausgeschieden. Proteine können enzymatisch an Ubiquitin gekoppelt werden (Ubiquitinierung) und
dadurch ihre Eigenschaften verändern; das dient Qualitätskontrolle,
Belastungsreaktionen, Regulierung des Zellzyklus, MHC-Präsentation,
oder Abbau (Proteasomen). Hormone haben Halbwertszeiten von wenigen Minuten bis zu mehreren Tagen Es
gibt verschiedene Wege, Wirkstoffe in den Körper zu bringen:
Nichtinvasive und invasive. Intraokular (Bindehaut), sublingual
(Schleimhaut von Zunge / Mundhöhle), bukkal (Wangenschleimhaut),
inhalativ / intranasal / intrapulmonal (Lunge), enteral (Schleimhaut
von Magen und Dünndarm), vaginal, rektal, transdermal, aural
(Gehörgang) sind Möglichkeiten, die Substanz über unverletzte Haut oder
Schleihäute in den Kreislauf zu bringen - über den Pfortaderkreislauf
primär zur Leber (enteral - dabei sind die Stoffe den
Verdauungsvorgängen ausgesetzt) oder direkt in den systemischen
Kreislauf. Wege unterschiedlicher Invasivität sind subkutan,
intravenös, intraarteriell, intramuskulär, intraartikulär (Gelenk),
intraperitoneal, intrakardial, epidural. Art und
Zeitverlauf der auf die Applikation folgenden Konzentrationsprofile
hängen von der Physiologie der Resorptionsprozesse ab Der Blutspiegel einer Substanz hängt ab von der im Kreislauf vorhandene Anfangsmenge, der durch Sekretion hinzukommenden und durch Speicherung, Abbau und Ausscheidung
verschwindenden Menge. Die Substanz kann mehr oder weniger intensiv an
Plasmaeiweiß gebunden sein; die gebundene Fraktion verbleibt
(weitgehend) im Kreislauf. Mit dem Anteil proteingebundenen Hormons
steigt seine Halbwertszeit. Die
Wirkung eines Signalstoffs hängt ab von seiner Konzentration am
Wirkungsort, der Ausstattung und Zugänglichkeit der Empfängerzellen mit
Rezeptoren, sowie den zwischen Rezeptor und Effekt geschalteten
Mechanismen Kinetik umfasst
Resorption, Transport, Distribution, Metabolisierung (Aktivierung /
Inaktivierung, Konjugation, Abbau) und Exkretion. Das Zeitprofil einer
Substanz nach ihrem Einbringen in den Kreislauf hängt von solchen
kinetischen Faktoren ab, z.B. der Permeabilität der Gefäßwände für
diesen Stoff. Die Verteilungskinetik lässt sich bei Kenntnis der wesentlichen Parameter - Bioverfügbarkeit, Verteilungsvolumen, Clearance, Halbwertszeit - mathematisch
modellieren. Komplexe Modelle berücksichtigen unterschiedliche
Eigenschaften verschiedener Organe und Gewebe. Die Art von
Proben, die für Konzentrationsmessungen herangezogen werden (Blut, Harn
etc), bestimmt auch die Art der Abbildung des Konzentrations- und Zeitverlaufs Dynamik
beschäftigt sich mit der Aktivität einer Wirksubstanz im Körper: Welche
Rezeptoren werden aktiviert? Welcher Mechanismus tritt anschließend in
Kraft (Membrankanäle, Enzyme, Gen(in)aktivierung)? Wie hängt die
biologische Wirkung von Gewebe, Dosis und Zeitverlauf ab? Peptidhormone werden enzymatisch abgebaut, Schilddrüsenhormone dejodiniert, Steroidhormone durch Biotransformation in wasserlöslichere Metabolite umgewandelt und diese mit Harn und Galle ausgeschieden. Proteine können enzymatisch an Ubiquitin gekoppelt werden (Ubiquitinierung) und
dadurch ihre Eigenschaften verändern; das dient Qualitätskontrolle,
Belastungsreaktionen, Regulierung des Zellzyklus, MHC-Präsentation,
oder Abbau (Proteasomen). Hormone haben Halbwertszeiten von wenigen Minuten bis zu mehreren Tagen Es
gibt verschiedene Wege, Wirkstoffe in den Körper zu bringen:
Nichtinvasive und invasive. Intraokular (Bindehaut), sublingual
(Schleimhaut von Zunge / Mundhöhle), bukkal (Wangenschleimhaut),
inhalativ / intranasal / intrapulmonal (Lunge), enteral (Schleimhaut
von Magen und Dünndarm), vaginal, rektal, transdermal, aural
(Gehörgang) sind Möglichkeiten, die Substanz über unverletzte Haut oder
Schleihäute in den Kreislauf zu bringen - über den Pfortaderkreislauf
primär zur Leber (enteral - dabei sind die Stoffe den
Verdauungsvorgängen ausgesetzt) oder direkt in den systemischen
Kreislauf. Wege unterschiedlicher Invasivität sind subkutan,
intravenös, intraarteriell, intramuskulär, intraartikulär (Gelenk),
intraperitoneal, intrakardial, epidural. Art und
Zeitverlauf der auf die Applikation folgenden Konzentrationsprofile
hängen von der Physiologie der Resorptionsprozesse ab
|
