

Eine Reise durch die Physiologie - Wie der Körper des Menschen funktioniert


 Kinetik und Regulation
Kinetik und Regulation
 (Ant)Agonist: αντι = gegen, άγω = ich handle, bewege
(Ant)Agonist: αντι = gegen, άγω = ich handle, bewege| Der Effekt eines Hormons / Wirkstoffs hängt u.a. von seiner Konzentration am Wirkort ("Dosis") ab. Das läßt sich mittels einer Dosis-Wirkungs-Kurve quantifizieren; sie kennzeichnet auch die Bindungskinetik zwischen Hormon und Rezeptor. Man erkennt eine maximale Wirkung (Emax) und die Hormondosis, welche 50% von Emax (halbmaximale Wirksamkeit) hervorruft. Die Affinität sagt aus, wie gut ein Stoff an einen bestimmten Rezeptor bindet (das kann für ein Hormon, aber auch seinen Antagonisten gelten); die Effizienz quantifiziert den biologischen Effekt (z.B. Signaltransduktion → zelluläre Antwort). Hormonrezeptoren in der Zellmembran werden oft nach ihrer Bindung internalisiert (endozytiert) und gehen dadurch der Signalwirkung ihres Hormons verloren: Die Zelle adaptiert, sie verliert ihre Empfindlichkeit (receptor downregulation): Die Zelle wird dem Hormon gegenüber (vorübergehend) refraktär - ganz oder teilweise. Hormone haben sehr unterschiedliche biologische Halbwertszeiten: Einige werden innerhalb weniger Minuten weitgehend aus dem Kreislauf entfernt (z.B. Angiotensin, Adrenalin); bei einigen braucht es bis zu einer halben Stunde, bis eine sezernierte Menge im Blut halbiert ist (z.B. Aldosteron, Prolaktin); wieder andere verbleiben für mehrere Stunden (einige Vorderlappenhormone); Schilddrüsenhormone sind so stark proteingebunden, dass ihre Halbwertszeit 1-7 Tage beträgt. |
 Regulierung und Modulation Biologische Verfügbarkeit
Neuronale und endokrine Kontrolle
Regulierung und Modulation Biologische Verfügbarkeit
Neuronale und endokrine Kontrolle
 Biologische Halbwertszeit
Biologische Halbwertszeit

 Abbildung:
Agonist und Antagonist
Abbildung:
Agonist und Antagonist
Agonisten  sind Substanzen, die durch Bindung an Rezeptoren die zelluläre Signaltransduktion aktivieren. sind Substanzen, die durch Bindung an Rezeptoren die zelluläre Signaltransduktion aktivieren.Antagonisten hemmen den betreffenden Agonisten in seiner Wirkung am Rezeptor. |
ist die entscheidende Größe (z.B.
welche Änderung des Blutdrucks löst eine bestimmte Dosis Adrenalin
aus) und erklärt sich im Allgemeinen nicht nur aus einer einfachen
Rezeptorkinetik, sondern es interagieren dabei mehrere physiologische Faktoren
(Herzminutenvolumen, Gefäßkontraktion, blutdruckwirksame Reflexe u.a.).
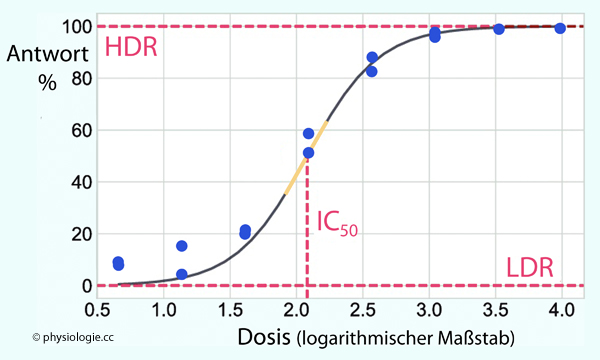
 Abbildung: Beispiel einer Dosis-Wirkungs-Kurve des Wirkstoffs, welche genau die Hälfte davon
(50%) hervorruft, können in vitro bestimmt werden. So kann z.B. die
Wirksamkeit unterschiedlicher Stoffe quantitativ verglichen werden.
Abbildung: Beispiel einer Dosis-Wirkungs-Kurve des Wirkstoffs, welche genau die Hälfte davon
(50%) hervorruft, können in vitro bestimmt werden. So kann z.B. die
Wirksamkeit unterschiedlicher Stoffe quantitativ verglichen werden. Die Bindung eines Wirkstoffs an einen Rezeptor bedeutet noch nicht,
dass diese auch eine Wirkung an der Zelle auslöst. Man unterscheidet Affinität, mit der die Bindung an den Rezeptor stattfindet, von Effizienz ,
mit der der biologische Effekt (Signaltransduktion → zelluläre Antwort) ausgelöst wird.
Die Bindung eines Wirkstoffs an einen Rezeptor bedeutet noch nicht,
dass diese auch eine Wirkung an der Zelle auslöst. Man unterscheidet Affinität, mit der die Bindung an den Rezeptor stattfindet, von Effizienz ,
mit der der biologische Effekt (Signaltransduktion → zelluläre Antwort) ausgelöst wird.  Variation der Rezeptorreserve verändert
die Empfindlichkeit der Zelle gegenüber dem betreffenden Signalstoff.
Variation der Rezeptorreserve verändert
die Empfindlichkeit der Zelle gegenüber dem betreffenden Signalstoff.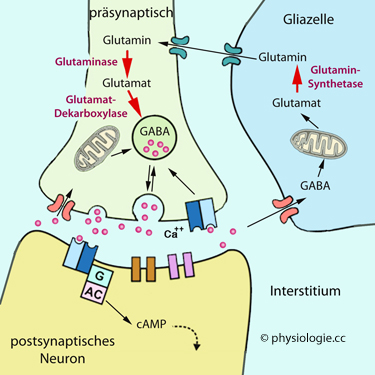 Abbildung: Wiederverwertung von GABA (transmitter uptake & release)
Abbildung: Wiederverwertung von GABA (transmitter uptake & release) cAMP, zyklisches Adenosinmonophosphat
G, G-Protein
GABA, γ-AminobuttersäureAbbildung). Die Bioverfügbarkeit
(bioavailability) einer Substanz gibt an, wie groß der Anteil der (im Körper vorhandenen)
Menge ist, der auch tatsächlich wirksam wird (vgl. dort). In der
Pharmakologie sagt man, die Bioverfügbarkeit eines i.v. gegebenen
Medikaments beträgt unmittelbar nach seiner Gabe 100% (der applizierten
Dosis). Die Bioverfügbarkeit
kann von zahlreichen Faktoren abhängen, wie tageszeitlichen Einflüssen,
Verteilungsmechanismus, Interaktion mit anderen Substanzen,
Biotransformation, Abbau und Ausscheidung (biologische Halbwertszeit!).
cAMP, zyklisches Adenosinmonophosphat
G, G-Protein
GABA, γ-AminobuttersäureAbbildung). Die Bioverfügbarkeit
(bioavailability) einer Substanz gibt an, wie groß der Anteil der (im Körper vorhandenen)
Menge ist, der auch tatsächlich wirksam wird (vgl. dort). In der
Pharmakologie sagt man, die Bioverfügbarkeit eines i.v. gegebenen
Medikaments beträgt unmittelbar nach seiner Gabe 100% (der applizierten
Dosis). Die Bioverfügbarkeit
kann von zahlreichen Faktoren abhängen, wie tageszeitlichen Einflüssen,
Verteilungsmechanismus, Interaktion mit anderen Substanzen,
Biotransformation, Abbau und Ausscheidung (biologische Halbwertszeit!). 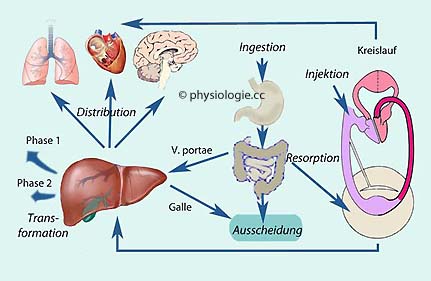 Abbildung: Komponenten der Bioverfügbarkeit
Abbildung: Komponenten der Bioverfügbarkeit Struktur und Eigenschaften des Verteilungsraums (Organismus)
Struktur und Eigenschaften des Verteilungsraums (Organismus) in den Kreislauf (aus z.B. einer Drüse) abgegebene / (von außen) eingebrachte Dosis oder Infusionsrate (maximale) Konzentration im Blut, Zeitpunkt des Erreichens der maximalen Konzentration Verteilungsvolumen / Verteilungsvolumina Clearance Bioverfügbarkeit der betrachteten Substanz Eine mit der Bioverfügbarkeit verknüpfte Kenngröße ist die biologische Halbwertszeit
(half life) der
betreffenden Moleküle (z.B. Hormone, Mediatoren, Transmitter), das
ist die Zeit, die vergeht, bis die zu einem Startzeitpunkt in den
Kreislauf eingebrachte Indikatormenge (des Stoffes, dessen
Halbwertszeit bestimmt werden soll) zur Hälfte
aus dem Blutkreislauf verschwunden und die Plasmakonzentration um 50%
reduziert ist.
in den Kreislauf (aus z.B. einer Drüse) abgegebene / (von außen) eingebrachte Dosis oder Infusionsrate (maximale) Konzentration im Blut, Zeitpunkt des Erreichens der maximalen Konzentration Verteilungsvolumen / Verteilungsvolumina Clearance Bioverfügbarkeit der betrachteten Substanz Eine mit der Bioverfügbarkeit verknüpfte Kenngröße ist die biologische Halbwertszeit
(half life) der
betreffenden Moleküle (z.B. Hormone, Mediatoren, Transmitter), das
ist die Zeit, die vergeht, bis die zu einem Startzeitpunkt in den
Kreislauf eingebrachte Indikatormenge (des Stoffes, dessen
Halbwertszeit bestimmt werden soll) zur Hälfte
aus dem Blutkreislauf verschwunden und die Plasmakonzentration um 50%
reduziert ist. Nach Injektion eines Bolus der (radioaktiv markierten) Biosubstanz
(Hormon, Metabolit, ..), wird diese im Körper oft kontinuierlich
abgebaut. Nicht bioaktive radioaktive Fragmente verfälschen die
Bestimmung.
Die Verteilung im Organismus (Kinetik) läuft in komplexer (oft mehrphasiger) Weise ab. Die Bindung an verschiedenen Stellen des Körpers (Dynamik)
erfolgt je nach Ausstattung der Zellen mit entsprechenden Rezeptoren.
In welchem Kompartiment (Blut, Plasma, Sekret, ..) und unter welchen Begleitumständen die Messungen erfolgt sind, kann sich auf das Resultat auswirken.
Nach Injektion eines Bolus der (radioaktiv markierten) Biosubstanz
(Hormon, Metabolit, ..), wird diese im Körper oft kontinuierlich
abgebaut. Nicht bioaktive radioaktive Fragmente verfälschen die
Bestimmung.
Die Verteilung im Organismus (Kinetik) läuft in komplexer (oft mehrphasiger) Weise ab. Die Bindung an verschiedenen Stellen des Körpers (Dynamik)
erfolgt je nach Ausstattung der Zellen mit entsprechenden Rezeptoren.
In welchem Kompartiment (Blut, Plasma, Sekret, ..) und unter welchen Begleitumständen die Messungen erfolgt sind, kann sich auf das Resultat auswirken. Hydrophile Signalstoffe (Peptide,
Aminosäurenabkömmlinge, Prostaglandine) sind gut im Blutplasma löslich
und benötigen typischerweise kein Transportvehikel (Ausnahmen
bestätigen die Regel - z.B. dienen IGF-Bindungsproteine der
Beeinflussung der IGF-Bioverfügbarkeit). Ihre Konzentration im Blutplasma
ist niedrig (10-12 bis 10-10 M/l). Sie gelangen leicht
an die Zellen im Gewebe, können jedoch die Zellmembran (Phospholipide!)
nicht durchdringen. Nach Bindung an Membranrezeptoren lösen solche Signalsubstanzen rasche
intrazelluläre Folgereaktionen (Minuten bis Stunden) aus
(
Hydrophile Signalstoffe (Peptide,
Aminosäurenabkömmlinge, Prostaglandine) sind gut im Blutplasma löslich
und benötigen typischerweise kein Transportvehikel (Ausnahmen
bestätigen die Regel - z.B. dienen IGF-Bindungsproteine der
Beeinflussung der IGF-Bioverfügbarkeit). Ihre Konzentration im Blutplasma
ist niedrig (10-12 bis 10-10 M/l). Sie gelangen leicht
an die Zellen im Gewebe, können jedoch die Zellmembran (Phospholipide!)
nicht durchdringen. Nach Bindung an Membranrezeptoren lösen solche Signalsubstanzen rasche
intrazelluläre Folgereaktionen (Minuten bis Stunden) aus
( s. dort).
Teils werden sie rezeptorgebunden von der Zelle aufgenommen, teils im
Blut, der Leber oder nach Filtration und Rückresorption in den
Nierentubuli proleolytisch abgebaut. Das erklärt eine kurze Halbwertszeit (typischerweise einige Minuten). Lipophile
(hydrophobe) Hormone benötigen Transportmoleküle, an die sie binden
(Löslichkeit) und die den Abbau durch Proteasen und renale Filtration
weitgehend verhindern. Das erhöht ihre Halbwertszeit (Stunden bis
Tage). Beispiele: Steroide, Schilddrüsenhormone. Diese Hormone
diffundieren - soferne sie bis ins Gewebe vorgedrungen sind - durch die
Zellmembran (da lipophil) und binden an intrazelluläre Rezeptoren.
Typischerweise beeinflussen sie dadurch die Proteinsynthese
(Transkription → Translation). Ihr Effekt setzt im Rahmen dieses zeitaufwendigen Mechanismus
zeitverzögert (1-2 Stunden) ein. s. dort).Abbildung).
Allen Regelsystemen ist gemeinsam, dass sie über ein Zentrum verfügen,
dass Zustandsvariablen misst und mit Sollwerten vergleicht
("integrierendes Zentrum"). Das können einzelne Zellen oder mehr oder
weniger komplexe Zellverbände sein, die dann meistens im
Zentralnervensystem zu finden sind.
s. dort).
Teils werden sie rezeptorgebunden von der Zelle aufgenommen, teils im
Blut, der Leber oder nach Filtration und Rückresorption in den
Nierentubuli proleolytisch abgebaut. Das erklärt eine kurze Halbwertszeit (typischerweise einige Minuten). Lipophile
(hydrophobe) Hormone benötigen Transportmoleküle, an die sie binden
(Löslichkeit) und die den Abbau durch Proteasen und renale Filtration
weitgehend verhindern. Das erhöht ihre Halbwertszeit (Stunden bis
Tage). Beispiele: Steroide, Schilddrüsenhormone. Diese Hormone
diffundieren - soferne sie bis ins Gewebe vorgedrungen sind - durch die
Zellmembran (da lipophil) und binden an intrazelluläre Rezeptoren.
Typischerweise beeinflussen sie dadurch die Proteinsynthese
(Transkription → Translation). Ihr Effekt setzt im Rahmen dieses zeitaufwendigen Mechanismus
zeitverzögert (1-2 Stunden) ein. s. dort).Abbildung).
Allen Regelsystemen ist gemeinsam, dass sie über ein Zentrum verfügen,
dass Zustandsvariablen misst und mit Sollwerten vergleicht
("integrierendes Zentrum"). Das können einzelne Zellen oder mehr oder
weniger komplexe Zellverbände sein, die dann meistens im
Zentralnervensystem zu finden sind. Abbildung: Strukturen neuronaler und endokriner Regulation Sie können einfach gestaltet sein, wie bei einfachen neuronalen (1,
links) oder endokrinen Reflexen (6, rechts); oder es handelt sich um zusammengesetzte
Funktionsmuster, bei denen einmal oder öfter ein Transportweg über die
Blutbahn integriert ist.
Abbildung: Strukturen neuronaler und endokriner Regulation Sie können einfach gestaltet sein, wie bei einfachen neuronalen (1,
links) oder endokrinen Reflexen (6, rechts); oder es handelt sich um zusammengesetzte
Funktionsmuster, bei denen einmal oder öfter ein Transportweg über die
Blutbahn integriert ist.

 Antagonisten konkurrieren mit Agonisten
um spezifische Bindungsstellen an Rezeptoren, an denen letztere eine
Konformationsänderung bewirken. Je höher die Konzentration des
Antagonisten, desto schwerer ist eine Wirkung durch den Agonisten zu
erzielen (kompetitive Hemmung). Stoffe, die an den Rezeptor binden,
ohne wirksam zu werden, hemmen den betreffenden Agonisten in seiner
Wirkung am Rezeptor. Die Dosis-Wirkungs-Kurve beschreibt die Beziehung zwischen Dosis und Effekt. Bei einer Responder-Fraktion von 0 ist die Dosis unterschwellig / wirkungslos; bei einer Fraktion von 1 reagieren alle Probenden (100%) Dosis-Wirkungs-Kurven quantifizieren die Bindungskinetik (Wirkstoff - Rezeptor). Man bestimmt die maximale Wirkung (Emax) und die Dosis des Wirkstoffs, welche genau die Hälfte davon hervorruft (EC50, ED50 - C = concentration, D = dose). Das ermöglicht den Vergleich der Wirksamkeit unterschiedlicher Agenzien. Affinität gibt das Maß der Bindung an den Rezeptor an, Effizienz
den biologischen Effekt (Signaltransduktion → zelluläre Antwort). Ein
Antagonist kann gute Affinität aufweisen, ohne Effizienz zu haben (z.B.
Rezeptorblocker). Einige Rezeptoren weisen auch ohne Bindung eines
Liganden Wirkung auf (Rezeptoren für Cannabinoide, Serotonin, einige
andere Mediatoren), sie sind sozusagen spontanaktiv ("konstitutiv aktiv") Bindet der
Agonist nur wenige (z.B. ~1-10%) der vorhandenen Rezeptoren, kann das
manchmal bereits eine volle biologische Wirkung erzielen, wie z.B. bei G-Protein-gekoppelten Neurotransmittern und Hormonen (die nicht an der Kopplung beteiligten Rezeptoren werden als Rezeptorreserve bezeichnet). Variationen
der Rezeptorreserve verändern die Empfindlichkeit der Zelle gegenüber
dem betreffenden Signalstoff. Wird die Zahl der Rezeptoren reduziert
(herunterreguliert), sinkt die Hormonbindungskapazität und damit
-wirkung. Zellen können auf diese Weise gegenüber Hormonen für einige
Zeit refraktär werden. Das kann durch rasche Aufnahme des Transmitters durch angrenzende Zellen verhindert werden, wie z.B. bei der Aufnahme von GABA durch Gliazellen im Kleinhirn Bioverfügbarkeit
quantifiziert den Anteil eines Stoffes, der tatsächlich wirksam ist.
Sie hängt von zahlreichen Faktoren ab (zirkadiane Einflüsse,
Verteilung, Interaktion mit anderen Substanzen, Biotransformation,
Abbau und Ausscheidung). Eine Kenngröße ist die biologische
Halbwertszeit. Hydrophile
Signalstoffe (Peptide, Aminosäurenderivate, Prostaglandine) sind gut im
Blutplasma löslich und benötigen kaum Transportproteine, ihre
Konzentration im Blutplasma ist niedrig, sie gelangen leicht an die
Zellen im Gewebe, binden an Membranrezeptoren und lösen rasche
intrazelluläre Folgereaktionen aus. Teils werden sie rezeptorgebunden
wiederaufgenommen, teils systemisch abgebaut: kurze Halbwertszeit. Lipophile
Signalstoffe benötigen Transportmoleküle, haben längere
Halbwertszeiten, diffundieren durch die Zellmembran, binden an
intrazelluläre Rezeptoren und beinflussen die Expression von Zielgenen;
ihr Effekt setzt zeitverzögert ein Antagonisten konkurrieren mit Agonisten
um spezifische Bindungsstellen an Rezeptoren, an denen letztere eine
Konformationsänderung bewirken. Je höher die Konzentration des
Antagonisten, desto schwerer ist eine Wirkung durch den Agonisten zu
erzielen (kompetitive Hemmung). Stoffe, die an den Rezeptor binden,
ohne wirksam zu werden, hemmen den betreffenden Agonisten in seiner
Wirkung am Rezeptor. Die Dosis-Wirkungs-Kurve beschreibt die Beziehung zwischen Dosis und Effekt. Bei einer Responder-Fraktion von 0 ist die Dosis unterschwellig / wirkungslos; bei einer Fraktion von 1 reagieren alle Probenden (100%) Dosis-Wirkungs-Kurven quantifizieren die Bindungskinetik (Wirkstoff - Rezeptor). Man bestimmt die maximale Wirkung (Emax) und die Dosis des Wirkstoffs, welche genau die Hälfte davon hervorruft (EC50, ED50 - C = concentration, D = dose). Das ermöglicht den Vergleich der Wirksamkeit unterschiedlicher Agenzien. Affinität gibt das Maß der Bindung an den Rezeptor an, Effizienz
den biologischen Effekt (Signaltransduktion → zelluläre Antwort). Ein
Antagonist kann gute Affinität aufweisen, ohne Effizienz zu haben (z.B.
Rezeptorblocker). Einige Rezeptoren weisen auch ohne Bindung eines
Liganden Wirkung auf (Rezeptoren für Cannabinoide, Serotonin, einige
andere Mediatoren), sie sind sozusagen spontanaktiv ("konstitutiv aktiv") Bindet der
Agonist nur wenige (z.B. ~1-10%) der vorhandenen Rezeptoren, kann das
manchmal bereits eine volle biologische Wirkung erzielen, wie z.B. bei G-Protein-gekoppelten Neurotransmittern und Hormonen (die nicht an der Kopplung beteiligten Rezeptoren werden als Rezeptorreserve bezeichnet). Variationen
der Rezeptorreserve verändern die Empfindlichkeit der Zelle gegenüber
dem betreffenden Signalstoff. Wird die Zahl der Rezeptoren reduziert
(herunterreguliert), sinkt die Hormonbindungskapazität und damit
-wirkung. Zellen können auf diese Weise gegenüber Hormonen für einige
Zeit refraktär werden. Das kann durch rasche Aufnahme des Transmitters durch angrenzende Zellen verhindert werden, wie z.B. bei der Aufnahme von GABA durch Gliazellen im Kleinhirn Bioverfügbarkeit
quantifiziert den Anteil eines Stoffes, der tatsächlich wirksam ist.
Sie hängt von zahlreichen Faktoren ab (zirkadiane Einflüsse,
Verteilung, Interaktion mit anderen Substanzen, Biotransformation,
Abbau und Ausscheidung). Eine Kenngröße ist die biologische
Halbwertszeit. Hydrophile
Signalstoffe (Peptide, Aminosäurenderivate, Prostaglandine) sind gut im
Blutplasma löslich und benötigen kaum Transportproteine, ihre
Konzentration im Blutplasma ist niedrig, sie gelangen leicht an die
Zellen im Gewebe, binden an Membranrezeptoren und lösen rasche
intrazelluläre Folgereaktionen aus. Teils werden sie rezeptorgebunden
wiederaufgenommen, teils systemisch abgebaut: kurze Halbwertszeit. Lipophile
Signalstoffe benötigen Transportmoleküle, haben längere
Halbwertszeiten, diffundieren durch die Zellmembran, binden an
intrazelluläre Rezeptoren und beinflussen die Expression von Zielgenen;
ihr Effekt setzt zeitverzögert ein |
