Übersicht  Renin und juxtaglomerulärer Apparat Angiotensin und Angiotensinrezeptoren
Renin und juxtaglomerulärer Apparat Angiotensin und Angiotensinrezeptoren

 Juxtaglomerulärer Apparat. macula densa
Juxtaglomerulärer Apparat. macula densa
 Praktische Aspekte
Praktische Aspekte  Core messages
Core messages
Die
Nieren stehen in einem Knotenpunkt der Regulierung von
Körperflüssigkeiten und Kreislauffunktion. Unter ihren Möglichkeiten,
auf diese Systeme einzuwirken, ist Renin
besonders bedeutsam: Bei drohendem Blutdruckabfall schütten die Nieren
dieses Enzym in das Blut aus. Renin setzt dann aus einem Plasmaprotein
Angiotensin frei, das im Kreislauf in Angiotensin II
umgewandelt wird. Dieses bewirkt ein umfangreiches Ensemble
blutdrucksteigernder Effekte: Vasokonstriktion, Durstempfinden,
gesteigerte Sekretion von Noradrenalin aus sympathischen Nervenfasern,
Freisetzung des "Natriumsparers" Aldosteron aus den Nebennieren.
Das Renin-Angiotensin-Aldosteron-System zielt auf Kreislaufregulation und Elektrolythaushalt ab
Voraussetzung
für die Stabilität des Kreislaufs ist die
Aufrechterhaltung funktionsadäquater Beträge von Blutvolumen,
Förderfunktion des Herzens und der Tonisierung der Blutgefäße. Diese
Faktoren stehen unter dem Einfluss des Reninsystems: Dieses unterstützt
Wirkungen des Sympathikus, regt die Aldosteronausschüttung an und hilft
bei der Regulierung des Elektrolyt- und Volumenhaushalts.
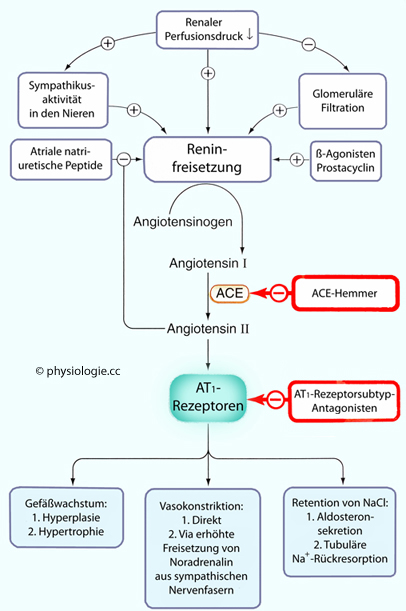
 Abbildung: Aufbau und Beeinflussung des Reninsystems
Nach einer Vorlage in Ritter / Flower / Henderson / Loke / MacEwan / Rang, Rang & Dale's Pharmacology, 9th ed. Elsevier 2020
Abbildung: Aufbau und Beeinflussung des Reninsystems
Nach einer Vorlage in Ritter / Flower / Henderson / Loke / MacEwan / Rang, Rang & Dale's Pharmacology, 9th ed. Elsevier 2020
Zu den Reizen,
welche die Freisetzung von Renin anregen, gehören Reduktion von
Perfusionsdruck und glomerulärer Filtration in der Niere, Abfall der Na+-Konzentration
im distalen Tubulus, verstärkte adrenerge Aktivität, Prostacyclin.
Angiotensin hemmt die Reninproduktion über Inhibition natriuretischer
Peptide (negatives Feedback).
Angiotensin I (10 AS) ist so gut wie wirkungslos, es wird durch Aktivität des endothelialen Enzyms ACE (angiotensin converting enzyme)
zum Octapeptid Angiotensin II. ACE wirkt vor allem in der Lunge (sehr
große Oberfläche des pulmonalen Kapillarbetts - entspricht der Fläche
eines halben Fußballfeldes), aber auch in anderen Gefäßsystemen
(Gehirn, Nieren, Herzmuskel, Skelettmuskulatur).
Die Wirkungen von Angiotensin II auf den AT1-Rezeptor- Subtyp schließen ein: Vasokonstriktion (insbesondere an vasa efferentia der Niere → verstärkte glomeruläre Filtration), vermehrte tubuläre Rückresorption von Na+, Aldosteronsekretion in der Nebenniere, forcierte Freisetzung von Noradrenalin aus sympathischen Varikositäten, Anregung des Wachstums von Herzmuskel- und Gefäßwandzellen.
ACE-Hemmer und AT1-Rezeptorantagonisten senken den Blutdruck
 Beispielsweise reagieren die Nieren auf erniedrigten Blutdruck mit der Produktion des Enzyms Renin
Beispielsweise reagieren die Nieren auf erniedrigten Blutdruck mit der Produktion des Enzyms Renin . D
. Dieses
wird in
juxtaglomerulären (granulären) Zellen
am vas afferens - zusammen mit der Vorstufe Prorenin - synthetisiert,
gespeichert und bei Aktivierung in die Blutbahn
freigesetzt (

Abbildung
unten).
Die Freisetzung von Renin wird durch folgende Faktoren gefördert:

Sinkende Strömung durch das tubuläre System, gemessen an der macula densa
Niedriger Blutdruck im vas afferens (granuläre Zellen wirken als intrarenale Barorezeptoren)
Sympathische Anregung (über ß
1-Rezeptoren)
 Die Aktivierung des Reninsystems erfolgt durch Kochsalzmangel, (zentralen) Blutdruckabfall und / oder Hypovolämie.
Die Aktivierung des Reninsystems erfolgt durch Kochsalzmangel, (zentralen) Blutdruckabfall und / oder Hypovolämie.
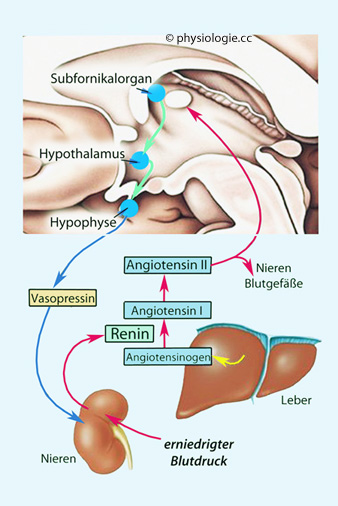
Abbildung: Interaktion Gehirn - Nieren
Nach einer Vorklage in Baer / Connors / Paradiso, Neuroscience. Exploring the Brain. 4th ed. Wolters Kluwer 2016
Abfall von Blutvolumen oder Blutdruck stimuliert die Niere zur Reninproduktion. Angiotensin reizt Zellen des Subfornikalorgans, dessen Neurone regen den Hypothalamus an, was zur Ausschüttung von Vasopressin und Durstempfinden führt.
Das ventral
des Fornix gelegene Subfornikalorgan
hat
angiotensinempfindliche Neurone und beteiligt sich an der Kontrolle des
Salz- und Wasserhaushalts. Erregung seiner Neurone erzeugt unmittelbar
Durstgefühl, das bei Inhibition dieser Neurone sofort aufhört
 Östrogenhaltige Kontrazeptiva können den Blutdruck auf diesem Wege
steigern: Östrogen ↑ → erhöhtes Angiotensinogen → AT II ↑→
Blutdruckanstieg.
Das endotheliale zinkhältige angiotensinkonvertierende Enzym (Angiotensin-Converting Enzyme ACE = Kininase II) spaltet davon zwei
Aminosäuren ab, es entsteht das stark blutdrucksteigernde Angiotensin
II.
Dieses wirkt nicht nur vasokonstriktorisch, sondern auch rückkoppelnd
auf das Gehirn (Abbildung): Es regt Durstempfinden und Salzappetit
an (Steigerung der Salzaufnahme erhöht das extrazelluläre
Flüssigkeitsvolumen), und regt die Vasopressininkretion an (verstärkte
Rückresorption von Wasser). Beides stabilisiert das Blutvolumen.
Östrogenhaltige Kontrazeptiva können den Blutdruck auf diesem Wege
steigern: Östrogen ↑ → erhöhtes Angiotensinogen → AT II ↑→
Blutdruckanstieg.
Das endotheliale zinkhältige angiotensinkonvertierende Enzym (Angiotensin-Converting Enzyme ACE = Kininase II) spaltet davon zwei
Aminosäuren ab, es entsteht das stark blutdrucksteigernde Angiotensin
II.
Dieses wirkt nicht nur vasokonstriktorisch, sondern auch rückkoppelnd
auf das Gehirn (Abbildung): Es regt Durstempfinden und Salzappetit
an (Steigerung der Salzaufnahme erhöht das extrazelluläre
Flüssigkeitsvolumen), und regt die Vasopressininkretion an (verstärkte
Rückresorption von Wasser). Beides stabilisiert das Blutvolumen.
Angiotensin II wirkt nicht nur vasokonstriktorisch, sondern ist auch indirekt kreislaufwirksam:
Es wirkt positiv inotrop auf Kardiomyozyten (erhöhte Herzleistung),
erhöht die Natrium-Rückresorption in der Niere (Steigerung des
extrazellulären Volumens), regt die Aldosteronbildung in der
Nebennierenrinde an, stimuliert das Durstempfinden (erhöhte
Wasseraufnahme), erleichtert die Freigabe von Noradrenalin aus
postganglionären Fasern, und wirkt als Wachstumsfaktor trophisch auf
das Herzmuskelgewebe.
Neurale und
hormonelle Mechanismen unterstützen die osmo-
und volumenregulatorische Einstellung des Gleichgewichts Salzaufnahme
- Salzausscheidung.
Die Systeme der Kreislauf-, Volumen-,
Elektrolyt- und Nierenregulation
sind daher eng verknüpft, und das Renin- Angiotensin- Aldosteron- Systems ist eine zentrale Säule für die Aufrechterhaltung des
Blutdrucks.
 Im Ruhezustand ist der Blutspiegel an Angiotensin II nicht hoch genug,
um kreislaufwirksam zu sein. Nach Blutverlust, bei körperlicher
Belastung, orthostatischer Herausforderung oder in ähnlichen
Situationen - die den Sympathikustonus erhöhen - hingegen wird das
Reninsystem aktiviert,
der Angiotensinspiegel steigt an. Die Folge ist Vasokonstriktion im
Splanchnikusgebiet und in den Nieren, was die Reninbildung noch weiter
steigert. Bei übertriebener Aktivierung dieses Mechanismus kann das positive Feedback zu akutem Nierenversagen führen.
Im Ruhezustand ist der Blutspiegel an Angiotensin II nicht hoch genug,
um kreislaufwirksam zu sein. Nach Blutverlust, bei körperlicher
Belastung, orthostatischer Herausforderung oder in ähnlichen
Situationen - die den Sympathikustonus erhöhen - hingegen wird das
Reninsystem aktiviert,
der Angiotensinspiegel steigt an. Die Folge ist Vasokonstriktion im
Splanchnikusgebiet und in den Nieren, was die Reninbildung noch weiter
steigert. Bei übertriebener Aktivierung dieses Mechanismus kann das positive Feedback zu akutem Nierenversagen führen.
Ein zweiter Garant der Kreislaufstabilität ist die Steuerung der kardialen Vorlast. Nimmt die
diastolische Füllung des Herzens ab, steigt reflektorisch die Aktivität des Sympathikus. Ziel ist eine
Stärkung der Herztätigkeit, arterioläre Vasokonstriktion (erhöhter
peripherer Widerstand) und Bereitstellung von Blut aus dem Niederdrucksystem.
Außer dem geschilderten hepatisch-renalen, systemisch wirkenden Reninsystem gibt es auch ein lokales (Gewebe-) Renin-Angiotensin-System. Zellen
in Gehirn, Nieren und Nebennieren, Fettgewebe, Intestinaltrakt,
Blutgefäßen und im Herzen
exprimieren Renin, Angiotensinogen, Angiotensine und / oder ACE. Dieses
System verstärkt die Wirkung systemisch erhöhter Angiotensinspiegel,
reguliert Perfusion und zelluläre Proliferationsvorgänge.
Die Aldosteronfreisetzung aus der Nebenniere kann auf drei Wegen
angeregt werden: Reninsystem (Nieren), ACTH (Gehirn) und erhöhter
Kaliumspiegel (Abbildung).

Abbildung: Renin- Angiotensin- Aldosteron- System
Nach einer Vorlage in Boron / Boulpaep: Concise Medical Physiology, Elsevier 2021
Die
drei Faktoren, welche die zona glomerulosa der Nebennierenrinde zur
Sekretion von Aldosteron anregen, sind der Renin-Angiotensin-Weg, ACTH
aus dem Hypophysenvorderlappen und ein erhöhter Kaliumspiegel im
Blutplasma.
Steigender Kaliumspiegel im Extrazellulärraum stimuliert Zellen der
zona glomerulosa der Nebennierenrinde zur Freisetzung von Aldosteron,
dieses führt in den renalen Sammelrohren zu vermehrter Rückresorption
von Natrium und Sekretion von Kalium.
Blockade
der Aldosteronproduktion kann zu lebensbedrohlicher Hyperkaliämie
führen. Hyperaldosteronismus bedingt gesteigerte Natriumresorption,
erhöht das extrazelluläre Volumen und ist für jeden zehnten Fall von
Bluthochdruck verantwortlich
Auch spielen alltägliche akute Veränderungen des Blutangebots an das Herz eine Rolle: Jeder Wechsel von liegender zu aufrechter Körperlage führt zu einer
solchen Situation: Die diastolische Füllung des Herzens (Vorlast) nimmt
ab, das Herz erhält weniger
Blut für seine Pumpfunktion (z.B. statt 7 l/min nur 5 l/min). Die Folge:
 Kardiopulmonale
Kardiopulmonale
Dehnungsrezeptoren ("
Volumenrezeptoren") werden weniger stark gedehnt.
Umgekehrt werden diese Rezeptoren bei erhöhter Belastung (Hinlegen,
plötzliche Hypervolämie) aktiviert; dadurch kommt es zu Hemmung des
Sympathikus und u.a. reduzierte Reninfreisetzung.
Dehnung von Volumenrezeptoren (Vorhöfe des Herzens) senkt reflektorisch die Freisetzung von Renin in den Nieren sowie von Vasopressin aus der Hypophyse (→ vermehrte Diurese)
|
Der
Karotissinus wird beim Aufrichten des Körpers ebenfalls schwächer gedehnt.
Über reduzierte Aktivität vagaler Afferenzen zum nucl. tractus
solitarii, negative Rückkopplung, Anregung des Sympathikus (Barorezeptorreflex) werden Herzfrequenz und
Gefäßmuskeltonus gesteigert. So kann der Blutdruck trotz geringerer Auswurfleistung des Herzens stabil gehalten werden.
Dazu kommen die Wirkungen hormoneller Systeme, vor allem des Renin-Angiotensin-Aldosteron-Systems.
Renin
Der juxtaglomeruläre Apparat (JGA, juxtaglomerular apparatus) ist eine Gruppe von Zellen zwischen Nierentubulus und dem zugehörigen vas afferens.
Diese Zellen detektieren Veränderungen der tubulären
Flüssigkeitsströmung und beeinflussen dementsprechend den Tonus des vas
afferens. Das ermöglicht tubulo-glomeruläre Rückkopplung, d.h. die Beeinflussung der glomerulären Filtration durch die Strömung im Tubulus. Die macula densa ist
eine Gruppe modifizierter Tubulusepithelzellen im Bereich des
juxtaglomerulären Apparats, die sich am tubulo-glomerulären Feedback
beteiligen ( Abbildung).

Abbildung: Juxtaglomerulärer Apparat
Modifiziert nach einer Vorlage in Boron / Boulpaep: Concise Medical Physiology, Elsevier 2021
Der juxtaglomeruläre Apparat steuert das Reninsystem; granuläre Zellen - spezielle Wandzellen des vas afferens - bilden Renin. Granuläre Zellen werden von sympathischen Fasern innerviert, diese regen die Reninsekretion ß-adrenerg an.
Zellen der macula densa (in der Wand des frühen distalen Tubulus, der
mit dem Mesangium Kontakt aufnimmt) reagieren auf Veränderungen der
Kochsalzkonzentration im Tubulus, indem sie die Reninsekretion und den Tonus der Wand des vas afferens beeinflussen. Resultat
ist eine Erhöhung der Filtration und des Blutdrucks.
Mesangiumzellen
sezernieren extrazelluläre Matrix und sind kontraktil. Sie reagieren
auf vasoaktive Stoffe und kommunizieren mit Podozyten (molekular) -
dadurch beeinflussen sie die
Filtration - sowie mit glatten Muskelzellen (via gap junctions). Auf
Dehnung der Kapillarwände bilden sie verschiedene Wachstumsfaktoren
Der juxtaglomeruläre Apparat besteht aus extraglomerulären mesangialen Zellen
(auch Goormaghtigh- , Polkissen- oder Lacis-Zellen),
granulären Zellen des vas afferens, der macula densa und Teilen des aufsteigenden Tubulusschenkels.
Spezialisierte glattmuskuläre Wandzellen des vas afferens werden als juxtaglomeruläre oder granuläre Zellen bezeichnet. Sie kommunizieren über den lokalen Extrazellulärraum mit Zellen der macula densa.
Eine Erhöhung des Angiotensinspiegels (ATII) hemmt direkt die Reninsekretion der granulären Zellen (short-loop feedback); und indirekt durch ihre blutdrucksteigernde Wirkung.
 Eine Abnahme des effektiven zirkulierenden Blutvolumens bzw. des systemischen arteriellen Drucks triggert die Reninfreisetzung aus dem juxtaglomerulären Apparat auf drei Wegen:
Eine Abnahme des effektiven zirkulierenden Blutvolumens bzw. des systemischen arteriellen Drucks triggert die Reninfreisetzung aus dem juxtaglomerulären Apparat auf drei Wegen:
Verringerte Reizung arterieller Barorezeptoren bewirkt reflektorisch Erhöhung des Sympathikuseinflusses auf die Niere
Kochsalzkonzentration an der macula densa (NaCl-Sensor) nimmt ab
Sinkender Perfusionsdruck im Bereich der vasa afferentia entlastet Dehnungsrezeptoren (granuläre Zellen)
 Granuläre (juxtaglomeruläre) Zellen bilden (als Präprorenin, das über Prärenin zu Renin verwandelt
wird), speichern (vesikulär, die Freisetzung erfolgt bei Bedarf ohne
Verzögerung) und sezernieren
Renin.
Granuläre (juxtaglomeruläre) Zellen bilden (als Präprorenin, das über Prärenin zu Renin verwandelt
wird), speichern (vesikulär, die Freisetzung erfolgt bei Bedarf ohne
Verzögerung) und sezernieren
Renin.
Renin spaltet aus dem aus der Leber stammenden α2-Globulin Angiotensinogen das (kaum blutdruckwirksame) Dekapeptid Angiotensin I (ATI) ab. Angiotensin-Converting Enzyme (ACE) aus Endothelzellen macht aus ATI das
vasokonstriktorisch wirkende Oktapeptid Angiotensin II (ATII). Etwa 40% der ACE-Aktivität stammt aus dem Endothel des Lungenkreislaufs, der Rest /(~60%) verteilt sich auf die Endothelien des geasmten restlichen Körpers.
ATII erhöht den Blutdruck (auf äquimolarer Basis) etwa 40-mal so stark
wie Noradrenalin. Es kontrahiert hauptsächlich Gefäße in der Haut und
im Splachnikusgebiet (Organe mit starker venöser Blutspeicherkapazität
- bei Vasokonstriktion wird ein wesentlicher Anteil des Blutvolumens
von der Peripherie zum Herzen verlagert) sowie in den Nieren.
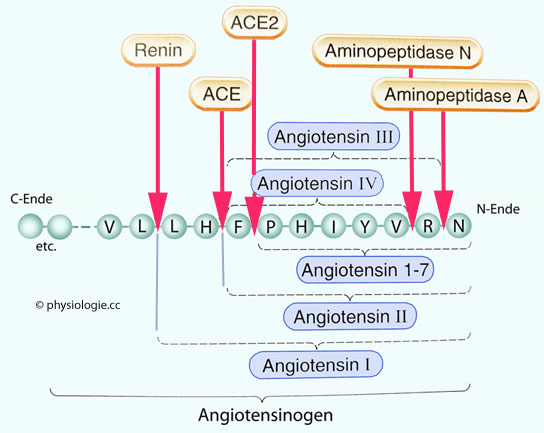
Abbildung: Enzyme und die Bildung von Angiotensin I bis IV aus Angiotensinogen
Nach einer Vorlage in Ritter / Flower / Henderson / Loke / MacEwan / Rang, Rang & Dale's Pharmacology, 9th ed. Elsevier 2020
Renin
spaltet AT I vom N-terminalen Ende des Angiotensinmoleküls. Durch
Einwirken von ACE entsteht daraus AT II, AT 1-7 resultiert aus Wirkung
von ACE2.
Aminopeptidase A verwandelt AT II in AT III, Aminopeptidase N verwandelt AT II in AT IV.
V = Valin, L = Leucin, H = Histidin, F = Phenylalanin, P = Prolin, I = Isoleucin, Y = Tyrosin, R = Arginin, N = Asparagin
Angiotensin II ist ein Substrat für mehrere Enzyme (Abbildung):
Außer für ACE auch für Aminopeptidase A (sie spaltet Asparagin vom
N-terminalen Ende ab, es entsteht Angiotensin III) und Aminopeptidase N
(Abspaltung von zwei Aminosäuren, es entsteht Angiotensin IV). Angiotensin III regt die Seklretion von Adosteron an; Angiotensin IV bewirkt - wahrscheinlich über einen eigenen Rezeptor - am Endothel die Freisetzung von Plasminogenaktivator- Inhibitor 1.
Vasodilatatorische Gegenkomponente: Eine Zink-Metalloproteinase, das als
Angiotensin-Converting Enzyme 2 (ACE2) bezeichnete Transmembranprotein, spaltet von Angiotensin II am Carboxy-Ende Phenylalanin ab und wandelt es zum vasodilatatorischen Angiotensin 1-7 um (Abbildung). Dadurch kann dieser Teil des Systems u.a. herzschonend wirken.
ACE2 findet sich vor allem in Zellen des gastrointestinalen Systems,
der Nieren und des Herzens (ein wenig auch an anderen Zellen, wie
zilientragenden Epithelzellen im Respirationstrakt - s. die folgende
Anmerkung). Es spaltet mehrere weitere Peptide (Bradykinin, Ghrelin,
Neurotensin u.a.). Angiotensin 1-7 kann auch auf anderem
enzymatischen Wege aus Angiotensin I entstehen.
 ACE2 wurde ursprünglich als Rezeptormolekül für SARS-CoV (severe acute respiratory syndrome coronavirus) entdeckt (2003). Es dient dem SARS-CoV2-Virus
als Rezeptor (Andockstruktur); mit ihm reagiert das virale
S-Glykoprotein (S1-Untereinheit), das auch die Fusion der Zellmembranen
(Virus und Wirtszelle) mediiert (S2-Untereinheit enthält ein fusion peptide).
ACE2 wurde ursprünglich als Rezeptormolekül für SARS-CoV (severe acute respiratory syndrome coronavirus) entdeckt (2003). Es dient dem SARS-CoV2-Virus
als Rezeptor (Andockstruktur); mit ihm reagiert das virale
S-Glykoprotein (S1-Untereinheit), das auch die Fusion der Zellmembranen
(Virus und Wirtszelle) mediiert (S2-Untereinheit enthält ein fusion peptide).
Angiotensin und Niere: Die macula densa
in der Wand der frühen pars convuluta des distalen Tubulus schmiegt
sich an das Polkissen des juxtaglomerulären Apparats. Granuläre Zellen reagieren vor allem auf einen Blutdruckabfall im
Gefäß, sowie auf Reize aus der macula densa:
Sie haben ß1-Rezeptoren, d.h. sympathische Fasern können die Reninfreisetzung anregen (ß-Blocker wirken auf diesem Weg stark antihypertensiv)
Vasa afferentia haben Barorezeptorfunktion: Erhöhter Blutdruck hemmt, verminderter Blutdruck stimuliert die Reninfreisetzung
Feedback-Schleife: Sinkt der Blutdruck,
dann nimmt auch die glomeruläre Filtration ab.
Vasodilatation im vas afferens und Konstriktion des vas efferens (über Angiotensin) wirkt dem entgegen und erhöht die Filtration; Aktivierung des Renin-Angiotensin-Aldosteron-Mechanismus steigert den Blutdruck.
Dieser Mechanismus wird bei anhaltender Reduktion des
Flüssigkeitsstroms durch das Nephron aktiviert und erfordert mehrere
Minuten, um zur Wirkung zu kommen.

Zum
tubulo-glomerulären Feedbackmechanismus s.
dort
Die Reninbildung
wird angeregt durch
 Sympathikusaktivität (ß-adrenerg) - der Sympathikus wird z.B. reflektorisch angeregt bei Blutdruckabfall und Hypovolämie (Blutverlust)
Abnahme des
Perfusionsdrucks in der Niere
sinkende Natriumkonzentration an der
macula densa (Angiotensin II und atriale natriuretische Peptide senken die Reninfreisetzung)
Aldosteronmangel oder Antagonisierung des Mineralcorticoidrezeptors
Sympathikusaktivität (ß-adrenerg) - der Sympathikus wird z.B. reflektorisch angeregt bei Blutdruckabfall und Hypovolämie (Blutverlust)
Abnahme des
Perfusionsdrucks in der Niere
sinkende Natriumkonzentration an der
macula densa (Angiotensin II und atriale natriuretische Peptide senken die Reninfreisetzung)
Aldosteronmangel oder Antagonisierung des Mineralcorticoidrezeptors
Renin wird bei mangelnder Aldosteronwirkung freigesetzt
Blutdrucksteigerung (z.B. durch Angiotensin II) hemmt die Reninsekretion
|
Über die Steuerung der Reninfreisetzung s. dort

Renin im Serum
s.
dort
PRA (Reninaktivität,
plasma renin activity)
s.
dort
Angiotensin-konvertierendes Enzym (ACE) s.
dort

Abbildung: Angiotensin-Converting Enzyme (ACE) ist identisch mit Kininase II
Nach einer Vorlage bei historischesarchiv.dgk.org
ACE
spaltet von Angioternsin I zwei Aminosäuren ab, es entsteht das stark
gefäßkonstringierende Dekapeptid Angiotensin II. Das Enzym spaltet gleichzeitig
Bradykinin, das vasodilatatorisch wirkt (Kinase II); dadurch wird die
Gefäßweite ebenfalls reduziert. Der steigende Gefäßwiderstand erhöht
den Blutdruck.
ACE-Hemmer wirken somit doppelt antihypertensiv: Sie reduzieren die
Bildung von Angiotensin II und hemmen gleichzeitig den Abbau
blutdrucksenkenden Bradykinins
Sinkende Aktivität des ACE vermindert die Bildung von ATII, der Blutdruck sinkt, es wird vermehrt Renin gebildet
|
Im Sinne einer negativen Rückkopplung hemmt Angiotensin II die Reninsekretion in der Niere (auch natriuretische Peptide hemmen die Reninsekretion, was z.T. ihre natriuretische Wirkung erklärt).
 Angiotensin II wird von der Leber inaktiviert, seine biologische Halbwertszeit beträgt
Angiotensin II wird von der Leber inaktiviert, seine biologische Halbwertszeit beträgt
~
20 Minuten.
Die Angiotensin-II-Bildung wird physiologisch gesteuert über die Reninbildung.

Reninbildung
reagiert auf Reize, die durch sinkendes Extrazellulärvolumen bedingt
sind: Druckabfall in den Nieren, Stimulierung renaler ß
1-Rezeptoren, Prostaglandinbildung.

Die Reninbildung wird gehemmt durch Blutdruckerhöhung, Einfluss durch Vagusimpulse, Atriopeptin.
Angiotensinrezeptor
Angiotensinrezeptoren existieren in zwei Subtyp-Formen, AT1- und AT2-Rezeptoren. Diese haben unterschiedliche Wirkungen:
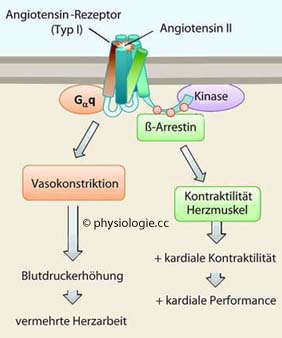
Abbildung: Angiotensinrezeptor
Nach DeWire SM, Violin JD. Biased Ligands for Better Cardiovascular Drugs - Dissecting G-Protein-Coupled Receptor Pharmacology. Circ Res 2011; 109: 205-16
Angiotensin
II aktiviert sowohl den G-Protein- als auch den ß-Arrestin-Mechanismus.
Ersterer führt zu Gefäßverengung und Blutdrucksteigerung, letzterer zu
erhöhter Kontraktilität des Herzmuskels
Der Angiotensin 1-Rezeptor (AT1-R) vermittelt
die Haupteffekte des Angiotensin II auf den Kreislauf. Er findet sich in
Blutgefäßen und anderen Geweben, und vermittelt Wirkungen wie Gefäßkontraktion, Retention von Flüssigkeit,
hyperplastisch-hypertrophe Effekte am kardiovaskulären System.
Die
Wirkung läuft über G-Proteine und vor allem Aktivierung der
Phospholipase C, weiters Hemmung der Adenylylcyclase und Aktivierung
der Phospholipase A2. Die Interaktion des Rezeptors mit G-Proteinen wird durch ß-Arrestine behindert (Desensitierung), die Rezeptoren werden in die Zelle internalisiert (downregulation);
und Arrestine aktivieren zusätzliche Signalwege, welche der Wirkung des
G-Protein-Signalweges entgegengesetzt sein können (Abbildung).
Die Wirkungen der AT1-Rezeptoren im Einzelnen:
Vasokonstriktion
(direkt sowie über verstärkte Noradrenalinfreisetzung aus sympathischen
Fasern), insbesondere an vasa efferentia, Haut- und Splanchnicusgefäßen
Abnahme der Nierendurchblutung,
Rückresorption von NaCl (sowohl über
Aldosteron als auch über erhöhte Aktivierung des Natrium-Wasserstoffionen-Austauscher
NHE am
proximalen Tubulus). Das steigert wiederum Plasmavolumen, venösen Rückstrom und
cardiac preload
(und arteriellen Blutdruck). Die
interstitielle Konzentration von Angiotensin in der Niere ist ~1000mal
höher als im Blutplasma, was auf eine starke parakrine
Wirkungskomponente hinweist
Aldosteronbildung, Vasopressinsekretion,
Verstärkung der Noradrenalinwirkung
Trophische Effekte auf Gefäße und
Herzmuskel
Der Angiotensin 2-Rezeptor (AT2-R) ist u.a. an der Mediierung der Apoptose beteiligt. Er wird vor allem in der Fetal- und Neonatalperiode exprimiert. Er regt die Produktion von NO an (vasodilatatorische Aktivität). Seine
physiologische Rolle ist teilweise unklar;
wahrscheinlich beeinflusst er Wachstums-, Differenzierungs-, Reifungs-
und Regenerierungsvorgänge.

Abbildung: Endokrine Blutdruckregulation
Nach Rossier BC, Bochud M, Devuyst O. The Hypertension Pandemic: An Evolutionary Perspective. Physiology 2017; 32: 112-25
Angiotensinwirkungen
an ihren Rezeptoren sind vermehrte Wasseraufnahme (Gehirn), erhöhte
glomeruläre Filtration und Salz-Wasser- Rückresorption (Nieren),
Anregung der Aldosteronsynthese (Nebennieren) und
Vasopressinfreisetzung (Hypophyse).
Diese Effekte wirken zusammen auf
den Kreislauf (Erhöhung von extrazellulärem und Blutvolumen - das
dauert einige Stunden - und totalem peripherem Widerstand - das geht
schnell - und damit Blutdruck).
Bei niedriger
Natriumzufuhr / Natriummangel nehmen Blutvolumen und Blutdruck ab, dies
stimuliert die Reninproduktion in der Niere und Anregung des RAAS. Es
folgt Stimulation von Angiotensinrezeptoren in Gehirn (Wasseraufnahme),
Nieren (Steigerung der glomerulären Filtration und der tubulären Salz-
und Wasserresorption), Nebennierenrinde (vermehrte Aldosteronbildung
und Natriumresorption) und im hypothalamisch-hypophysären System
(Vasopressinausschüttung, Wasserrückresorption). In Summe nehmen so
innerhalb einiger Stunden extrazelluläres Volumen und Blutdruck zu.
Vasokonstriktion der Widerstandsgefäße hingegen führt innerhalb weniger Minuten zu Blutdruckanstieg
AT II wird durch Peptidasen weiter verwandelt zu AT III (Durstempfinden,
Aldosteronfreisetzung) und AT IV (u.a. bremsender Einfluss auf
Fibrinolyseaktivität des Endothels über
Freisetzung des Plasminogen Activator Inhibitor-1). Rezeptoren für AT
IV finden sich auch im Hypothalamus. Dort erfolgt auch die Regulierung
von Durst und Trinkverhalten.
Angiotensin II: Wirkungsspektrum
AT II ist auf molarer Basis 10 bis
40-mal stärker vasokonstriktorisch aktiv als Noradrenalin (je nach Art
bzw. Rezeptorausstattung des Gefäßes). Seine Konzentration im Blut ist allerdings normalerweise gering, AT-II-Inhibitoren senken den Blutdruck unter physiologischen Umständen nur geringgradig.
Im Überblick: Angiotensin II wirkt
In der Niere vasokonstriktorisch auf vasa afferentia (über spannungsgesteuerte Calciumkanäle (VDCCs), die Ca++ in die Muskelzellen der Gefäßwände einströmen lassen) und (stärker) auf vasa efferentia
(vermutlich über Effekt auf die Phospholipase C (PLC), Aktivierung des
sarkoplasmatischen Retikulums durch Inositol-Triphosphat (IP3) und Steigerung des zytoplasmatischen [Ca++]).
Im Kreislauf vasokonstriktorisch, was den Blutdruck anhebt. Der Effekt erfolgt an der Gefäßwand über PLC, IP3, sarkoplasmatisches Retikulum und zytoplasmatisches [Ca++].
In den Nebennieren
stimuliert es die Freisetzung von Aldosteron, was zu einer Expansion
des Plasmavolumens führt und so den Kreislauf stabilisieren kann.
Im Gehirn fördert es das Durstempfinden (dipsogene Wirkung) durch Wirkung auf zirkumventrikuläre Organe
(area postrema, subfornikales Organ, vielleicht auch das organum
vasculosum laminae terminalis), und steigert die hypothalamische
Sekretion von Vasopressin (=Adiuretin), was das extrazelluläre Volumen vermehrt.
Im Fettgewebe scheint AT II das Gleichgewicht zugunsten der Lipogenese
(und Herabregulierung der Lipolyse) zu beeinflussen.
Angiotensin und Nebenniere: Physiologische ATII-Konzentrationen regen die
Aldosteronsekretion in der Nebennierenrinde an. ATII bindet an Typ-1 Angiotensin-II- Rezeptoren (AT1-Rezeptoren) in der Zellmembran von Zellen in der zona glomerulosa. Gq-vermittelt aktiviert dies die Phospholipase C (PLC), was DAG und IP3 entstehen lässt; DAG aktiviert Proteinkinase C und IP3 die Freisetzung von Ca++,
das Enzyme aktiviert, u.a. PKC und Proteinkinasen. Das führt zu einer
Depolarisierung der Zelle und anhaltenden Calciumeinstrom aus dem
Extrazellulärraum, wodurch die Sekretion von Aldosteron ansteigt (Ca++ regt die Produktion von Pregnenolon sowie die Aldosteronsynthase an)-
Auch
eine Erhöhung des extrazellulären Kaliumspiegels steigert den
Calciumeinstrom und damit die geschilderten Schritte zur Steigerung der
Aldosteronsekretion. Angiotensin II und Hyperkaliämie wirken auf
zona-glomerulosa-Zellen synergistisch, d.h. auf dieselben
intrazellulären Mechanismen.
Binnen etwa einer Stunde
steigt in der Niere die
Zahl der Na/K-ATPasen in der basolateralen, und der Na-Kanäle in der
apikalen Membran der Tubulusepithelzellen - Natrium wird resorbiert,
nimmt Wasser mit sich, stabilisiert Plasmavolumen und Blutdruck.
Angiotensin und Gehirn: Angiotensin II wirkt auf Hirnstamm (die area postrema
ist von der Blut-Hirn-Schranke ausgenommen und reagiert auf Angiotensin
mit einer Steigerung der Aktivität sympathischer Neurone),
sympathische Ganglien und Varikositäten (sie alle haben AT1-Rezeptoren)
im Sinne einer Vasokonstriktion. Angiotensin III steigert zusätzlich
das Durstempfinden (ein häufiges Symptom bei Hypovolämie), die Aufnahme
von Flüssigkeit hilft das Plasmavolumen zu erhöhen.
Angiotensin und Kreislauf: Besonders wichtige Effekte hat Angiotensin auf den peripheren Widerstand (TPR),
das renal-adrenale System sowie die Trophik des
kardiovaskulären Systems (Abbildung).

Abbildung: Hauptwirkungen von Angiotensin II
Nach einer Vorlage in Hilal-Dandan / Brunton, Goodman
& Gilman's Manual of Pharmacology and Therapeutics, 2nd ed., McGraw
Hill Education 2014
Bezüglich der Freisetzung von Aldosteron aus der
Nebennierenrinde sind AT II und AT III etwa gleich stark wirksam.
Im Rahmen pathologischer Vorgänge kann AT II über morphologische
Veränderungen im kardiovaskulären System (Hypertrophie) zu
blutdrucksteigernden Mechanismen beitragen
 Vasokonstriktion renaler und anderer Blutgefäße und Senkung der Perfusion. Kontraktion mesangialer Zellen bewirkt eine verringerte kapilläre Filtrationsfläche. Die Filtrationsrate nimmt bei niedrigem [AT II] ab,
bei hohen Konzentrationen kann sie aber durch prädominante Wirkung
am vas efferens zunehmen. ATII
reduziert andererseits den hydrostatischen Druck in peritubulären
Kapillaren und erhöht so die Rückresorption von Natrium und Flüssigkeit
im proximalen Tubulus.
Angiotensin II senkt noch auf einem weiteren Weg die Natrium- und Wasserausscheidung:
Vasokonstriktion renaler und anderer Blutgefäße und Senkung der Perfusion. Kontraktion mesangialer Zellen bewirkt eine verringerte kapilläre Filtrationsfläche. Die Filtrationsrate nimmt bei niedrigem [AT II] ab,
bei hohen Konzentrationen kann sie aber durch prädominante Wirkung
am vas efferens zunehmen. ATII
reduziert andererseits den hydrostatischen Druck in peritubulären
Kapillaren und erhöht so die Rückresorption von Natrium und Flüssigkeit
im proximalen Tubulus.
Angiotensin II senkt noch auf einem weiteren Weg die Natrium- und Wasserausscheidung:
Der vasokonstriktive Effekt senkt die Perfusion der vasa recta,
dadurch nimmt der Auswascheffekt für Harnstoff im Interstitium des Nierenmarks ab,
die steigende Harnstoffkonzentration führt zu vermehrter
NaCl-Resorption in der Henle-Schleife (aufsteigender Schenkel) -
Natriurese und Diurese sinken.
Verstärktes tubuloglomeruläres Feedback
durch erhöhte Sensitivität (intensiveres Absinken der glomerulären
Filtration bei gegebenem Anstieg des Natriums an der macula densa).
Verstärkter Na/H-Austausch im proximalen Tubulus, aufsteigenden Schenkel der Henle-Schleife sowie im oberen Sammelrohr. Angiotensin II in physiologischer Konzentration reguliert die Aktivität des Na+/H+-Austauschers Isoform 3 (NHE3) im
proximalen Tubulus über den Angiotensin-Rezeptor G-Protein-Mechanismus
hinauf.
Steigerung der Natriumresorption und Kaliumsekretion
(über Aldosteronwirkung), die Kochsalzausscheidungskurve als Funktion
des Blutdrucks wird insgesamt nach rechts (zu höheren Druckwerten hin)
verschoben.
Über längere Zeit bewirkt ATII Hypertrophie renaler Tubuluszellen.
Im Hypothalamus verstärkt ATII Durst und Vasopressinfreisetzung.
Eine der Wirkungen von Angiotensin II ist erhöhte Vasopressinausschüttung
|
Verstärkung der peripher-sympathischen Noradrenalinwirkung: AT II unterstützt die Freisetzung und hemmt die Wiederaufnahme von Noradrenalin an Nervenfaserendigungen.
Erhöhung des Sympathikustonus.
Freisetzung von Katecholaminen im Nebennierenmark durch Depolarisierung chromaffiner Zellen.
Förderung der Synthese und Sekretion von Aldosteron in der Nebennierenrinde, Verstärkung der sekretionsfördernden Wirkung von Kalium und ACTH.
Insgesamt unterstützt Angiotensin II Mechanismen, welche das effektive zirkulierende Volumen sowie den arteriellen Blutdruck stabilisieren oder erhöhen.
ACE-Hemmer
sind wichtige Blutdrucksenker. Eines davon ist Ramipril; dieses
antagonisiert nicht nur die Bildung von Angiotensin II, sondern fördert
auch den Abbau von Bradykinin. Bradykinin
wirkt vasodilatierend; daher bewirkt Ramipril eine Blutdrucksenkung
sowohl wegen der Senkung des AT II-spiegels als auch wegen des längeren
Verbleibs von Bradykinin an Gefäßwänden. Außerdem nimmt durch Reduktion
des AT II die Aldosteronkonzentration ab, was wiederum die Natriurese
fördert; die resultierende Verringerung des Plasmavolumens verstärkt
den blutdrucksenkenden Effekt des ACE-Antagonisten.
Angiotensinrezeptoren werden u.a. durch den AT1-Antagonisten Losartan gehemmt, der zur Behandlung von arterieller Hypertonie Verwendung findet.
Aldosteron wirkt über die Vermehrung aldosteronempfindlicher Natriumkanäle in der Zellmembran z.B. von Tubuluszellen (ENaC, s. auch dort). Kommt es zu Mutationen der ENaC, kann dies zu Pseudohypoaldosteronismus führen, wobei Hyperkaliämie und arterielle Hypotonie (Kochsalzmangel!) im Vordergrund stehen.
Insuffizienz der Nebennierenrinde (zona glomerulosa) hingegen bewirkt
Aldosteronmangel (Hypoaldosteronismus), der ähnliche Symptome
entwickelt (Mb. Addison).
Elektrolytveränderungen bei
renaler (renovaskulärer) Hypertonie: Eine Reduktion der
Nierendurchblutung (Stenose der a. renalis) aktiviert das Reninsystem
und führt zu erhöhten Aldosteronspiegeln. Folge ist vermehrte
Natriumretention und Hypernatriämie, gesteigerte Kaliumsekretion und Hypokaliämie sowie Alkalose infoge vermehrter H+-Ausscheidung.


 Richtig eingestelltes Blutvolumen ermöglicht stabilen Kreislauf über osmo- und volumenregulatorische
Einstellung des Gleichgewichts Salzaufnahme / Salzausscheidung (Natrium als extrazelluläres Leition). Cortisol,
Schilddrüsenhormone, Insulin, Östrogene (Blutdruckanstieg in der Schwangerschaft) regen in der Leber die Synthese des α2-Globulins Angiotensinogen an. Hypovolämie bzw. Blutdruckabfall aktivieren das Reninsystem: Renin setzt (als Peptidase) aus Angiotensinogen Angiotensin I frei. Endotheliales
angiotensinkonvertierendes Enzym (ACE = Kininase II) - vor allem in der Lunge - spaltet zwei
Aminosäuren ab, es entsteht stark blutdrucksteigerndes Angiotensin
II: Die Angiotensin-II-Bildung wird über die Reninbildung gesteuert Richtig eingestelltes Blutvolumen ermöglicht stabilen Kreislauf über osmo- und volumenregulatorische
Einstellung des Gleichgewichts Salzaufnahme / Salzausscheidung (Natrium als extrazelluläres Leition). Cortisol,
Schilddrüsenhormone, Insulin, Östrogene (Blutdruckanstieg in der Schwangerschaft) regen in der Leber die Synthese des α2-Globulins Angiotensinogen an. Hypovolämie bzw. Blutdruckabfall aktivieren das Reninsystem: Renin setzt (als Peptidase) aus Angiotensinogen Angiotensin I frei. Endotheliales
angiotensinkonvertierendes Enzym (ACE = Kininase II) - vor allem in der Lunge - spaltet zwei
Aminosäuren ab, es entsteht stark blutdrucksteigerndes Angiotensin
II: Die Angiotensin-II-Bildung wird über die Reninbildung gesteuert
Ständig verändert sich das Blutangebot an das Herz (Vorlast), vor allem bei Wechsel zwischen liegender und aufrechter Körperlage: Aufstehen senkt das Herzzeitvolumen akut um
~30%, kardiopulmonale und Carotis- Rezeptoren werden plötzlich weniger stark stimuliert, über reduzierte Aktivität vagaler Afferenzen zum nucl. tractus solitarii und negative Rückkopplung werden Sympathikustonus und Reninfreisetzung angeregt (Reninanstieg im Stehen). Die Aktivierung des Reninsystems stabilisiert Blutvolumen und venösen Rückstrom zum Herzen
Mesangiale, reninbildende granuläre (vas afferens) und Tubuluszellen (macula densa) bilden den juxtaglomerulären Apparat. Zellen in
der Wand des frühen distalen Tubulus registrieren die luminale
NaCl- Konzentration und beeinflussen Reninsekretion, Gefäßtonus und Blutdruck. Die Reninsekretion (aus Vesikeln in granulären Zellen) wird ß1-adrenerg, durch sinkende Natriumkonzentration an der macula densa, sowie Abnahme des renalen Perfusionsdrucks angeregt; Aktivierung des Reninmechanismus steigert den Blutdruck. Vasodilatation im vas afferens und Konstriktion des vas efferens (Angiotensin) erhöht die glomeruläre Filtration. Erhöhter Blutdruck hemmt, verminderter Blutdruck stimuliert die Reninfreisetzung
Angiotensin steigert die Aktivität sympathischer Neuronen im Hirnstamm (die area postrema ist von der Blut-Hirn-Schranke ausgenommen), stimuliert das Subfornikalorgan, was im Hypothalamus zur Ausschüttung von Vasopressin und Durstempfinden führt, und
regt die Aldosteronsekretion in der Nebennierenrinde an. Aldosteron
erhöht die Zahl der Na/K-ATPasen und der apikalen Na-Kanäle der renalen
Tubulusepithelzellen - Natrium wird resorbiert, nimmt Wasser mit sich,
stabilisiert Plasmavolumen und Blutdruck. Der Angiotensin 1-Rezeptor (AT1-R)
vermittelt die Haupteffekte des Angiotensin II auf den Kreislauf:
Vasokonstriktion (insbesondere Splanchnikusgebiet), Rückresorption von
NaCl / Sekretion von Kalium, Aldosteronbildung, Vasopressinsekretion, Verstärkung der Noradrenalinwirkung, Steigerung von extrazellulärem und Blutvolumen. Der Angiotensin 2-Rezeptor (AT2-R) wird vor allem in der Fetal- und Neonatalperiode exprimiert, wirkt vasodilatatorisch und ist an der Apoptose beteiligt
|
 Die Informationen in dieser Website basieren auf verschiedenen Quellen:
Lehrbüchern, Reviews, Originalarbeiten u.a. Sie
sollen zur Auseinandersetzung mit physiologischen Fragen, Problemen und
Erkenntnissen anregen. Soferne Referenzbereiche angegeben sind, dienen diese zur Orientierung; die Grenzen sind aus biologischen, messmethodischen und statistischen Gründen nicht absolut. Wissenschaft fragt, vermutet und interpretiert; sie ist offen, dynamisch und evolutiv. Sie strebt nach Erkenntnis, erhebt aber nicht den Anspruch, im Besitz der "Wahrheit" zu sein.
Die Informationen in dieser Website basieren auf verschiedenen Quellen:
Lehrbüchern, Reviews, Originalarbeiten u.a. Sie
sollen zur Auseinandersetzung mit physiologischen Fragen, Problemen und
Erkenntnissen anregen. Soferne Referenzbereiche angegeben sind, dienen diese zur Orientierung; die Grenzen sind aus biologischen, messmethodischen und statistischen Gründen nicht absolut. Wissenschaft fragt, vermutet und interpretiert; sie ist offen, dynamisch und evolutiv. Sie strebt nach Erkenntnis, erhebt aber nicht den Anspruch, im Besitz der "Wahrheit" zu sein.



 Renin, Angiotensin, Aldosteron
Renin, Angiotensin, Aldosteron
 Aldosteron: Aus "Aldehyd" und "Steroid"
Aldosteron: Aus "Aldehyd" und "Steroid"