



 Hämostase, Blutgerinnung, Fibrinolyse
Hämostase, Blutgerinnung, Fibrinolyse Fibrin, Fibronektin: fibra = Faser; nexus = Verknüpfung
Fibrin, Fibronektin: fibra = Faser; nexus = Verknüpfung| Bedingt durch Zustand und Eigenschaften der Gefäßwände werden im Blut gerinnungshemmende (antithrombotische) und -fördernde (prothrombotische)
Mechanismen aktiviert bzw. gehemmt. Auf diese Weise bleibt das Blut
einerseits (im Normalbetrieb) fließfähig, andererseits werden Schadstellen rasch verschlossen
(Blutstillung). Hauptakteure dieses fein abgestimmten komplexen Systems sind
Endothelzellen, Thrombozyten und plasmatische Faktoren der Gerinnung sowie die Fibrinolyse. Tritt eine Verletzung auf, gelangen Mechanismen in Aktion, die den Blutverlust gering halten. Zuerst kontrahieren sich Gefäße, was die Blutung mindert; verletzte Zellen fixieren und aktivieren Thrombozyten, die sich aneinanderhaften (Plättchen- oder weiße Thromben, beginnende Gefäßabdichtung) und zusammen mit der Gefäßwand das Gerinnungssystem aktivieren. In der Folge polymerisiert und vernetzt Fibrin (Vorstufe Fibrinogen: Faktor I). Im Fibrinnetz verfangen sich Blutkörperchen - hauptsächlich Erythrozyten (Gerinnungs- oder rote Thromben) - und helfen bei der Abdichtung des Gerinnsels. Innerhalb weniger Minuten steht mit etwas Glück die Blutung (Hämostase). Das Gerinnsel zieht sich schließlich zusammen (Retraktion). Unter dem Schutz dieser Abdichtung läuft die Wiederherstellung der Strukturen - die Wunde schließt sich, das Gewebe wird entweder vollständig (restitutio ad integrum) oder unter Zurückbleiben einer Narbe repariert. Das überflüssig gewordene Fibringerinnsel wird durch Fibrinolyse abgebaut: Die Serumprotease Plasmin wird durch Plasminaktivatoren freigesetzt und baut das Fibringerüst wieder ab. Plasminaktivatoren werden ihrerseits gehemmt, z.B. durch den endothelialen Plasminogenaktivator-Inhibitor. Der gesamte Mechanismus von Gerinnung und Fibrinolyse ist mehrfach auf sich selbst rückgekoppelt - ein Zeichen der Feinkontrolle, der dieses lebenswichtige System unterliegt. |
 Anti- vs. prothrombotische Eigenschaften Blutstillung Thrombozyten Gerinnungssystem Gerinnungskontrolle und Gerinnungshemmung Stabilisierung und Retraktion Fibrinolyse
Anti- vs. prothrombotische Eigenschaften Blutstillung Thrombozyten Gerinnungssystem Gerinnungskontrolle und Gerinnungshemmung Stabilisierung und Retraktion Fibrinolyse
 PAF Von Willebrand-Faktor (vWF) Vollblut, Serum Heparine Plasminogenaktivatoren
PAF Von Willebrand-Faktor (vWF) Vollblut, Serum Heparine Plasminogenaktivatoren
 Core messages
Core messages seine Fließfähigkeit einerseits
(je dünnflüssiger das Blut, desto besser für Transport
und kapillären Austausch), und die
Option zur Abdichtung verletzter Gefäße andererseits (hohe Viskosität
minimiert den Blutverlust an undichten Stellen - näheres zur Blutviskosität
seine Fließfähigkeit einerseits
(je dünnflüssiger das Blut, desto besser für Transport
und kapillären Austausch), und die
Option zur Abdichtung verletzter Gefäße andererseits (hohe Viskosität
minimiert den Blutverlust an undichten Stellen - näheres zur Blutviskosität  s. dort).
s. dort).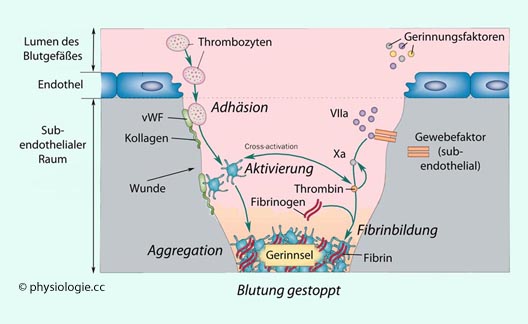
 Abbildung: Thrombozyten und Fibrin
Abbildung: Thrombozyten und Fibrin  als Hauptkomponenten der Blutstillung
als Hauptkomponenten der Blutstillung Der von Willebrand-Faktor (vWF) ist ein Glykoprotein, das von Endothelzellen (in Weibel-Palade- Körperchen),
subendothelialen Zellen sowie Megakaryozyten gebildet (von Thrombozyten
in α-Granula gespeichert) und an das Blutplasma abgegeben wird. Ein
vWF-Monomer enthält mehr als 2000 Aminosäuren und hat vielfache
Funktionen (bestimmte Domänen binden an Plättchen, Kollagen, Heparin,
Gerinnungsfaktor VIII).
Der von Willebrand-Faktor (vWF) ist ein Glykoprotein, das von Endothelzellen (in Weibel-Palade- Körperchen),
subendothelialen Zellen sowie Megakaryozyten gebildet (von Thrombozyten
in α-Granula gespeichert) und an das Blutplasma abgegeben wird. Ein
vWF-Monomer enthält mehr als 2000 Aminosäuren und hat vielfache
Funktionen (bestimmte Domänen binden an Plättchen, Kollagen, Heparin,
Gerinnungsfaktor VIII). Blut normalerweise nicht gerinnt und frei strömen kann (andernfalls Thrombosegefahr ), Blutungen jedoch rasch gestillt werden können (andernfalls Blutverlust und Schockgefahr).
Blut normalerweise nicht gerinnt und frei strömen kann (andernfalls Thrombosegefahr ), Blutungen jedoch rasch gestillt werden können (andernfalls Blutverlust und Schockgefahr).  Das Endothel (es bildet Faktoren wie den von Willebrand-Faktor, Prostazyklin und den gewebespezifischen Plasminogenaktivator t-PA): Gefäßkomponente Thrombozyten (sie sezernieren Stoffe für Plättchenaktivierung und Quervernetzung, Gerinnungsfaktoren, Calciumionen): Plättchenkomponente Das Koagulations- und Fibrinolysesystem (inkludiert gerinnungsfördernde und -hemmende sowie fibrinolysefördernde und -hemmende Substanzen): Gerinnungskomponente
Das Endothel (es bildet Faktoren wie den von Willebrand-Faktor, Prostazyklin und den gewebespezifischen Plasminogenaktivator t-PA): Gefäßkomponente Thrombozyten (sie sezernieren Stoffe für Plättchenaktivierung und Quervernetzung, Gerinnungsfaktoren, Calciumionen): Plättchenkomponente Das Koagulations- und Fibrinolysesystem (inkludiert gerinnungsfördernde und -hemmende sowie fibrinolysefördernde und -hemmende Substanzen): Gerinnungskomponente
 Abbildung: Erythrozyten in Fibrinnetz Abbildung).
Abbildung: Erythrozyten in Fibrinnetz Abbildung).
 Abbildung: Gerinnungshemmende (antithrombotische) Aktivitäten des Endothels
Abbildung: Gerinnungshemmende (antithrombotische) Aktivitäten des Endothels Antithrombotisch
(Abbildung): Normalerweise produzieren Endothelzellen Faktoren, die
das Aneinanderlagern von Blutplättchen und die Gerinnungsaktivität
hemmen, sowie die Auflösung (Lyse) allfälliger Thromben befördern. Eine
intakte Endothelschicht verhindert, dass Plättchen mit dem unter ihr
liegenden Bindegewebe (Basalmembran, Kollagenfasern etc) in Berührung
kommen. Membranassoziierte heparinähnliche Moleküle verstärken die Inaktivierung von Thrombin und anderen Gerinnungsfaktoren; Thrombomodulin auf der Oberfläche von Endothelzellen bindet Thrombin und verwandelt es in ein Antikoagulans, das Protein C aktiviert und dadurch die Gerinnungsfaktoren Va und VIIIa wirkungslos macht;
Antithrombotisch
(Abbildung): Normalerweise produzieren Endothelzellen Faktoren, die
das Aneinanderlagern von Blutplättchen und die Gerinnungsaktivität
hemmen, sowie die Auflösung (Lyse) allfälliger Thromben befördern. Eine
intakte Endothelschicht verhindert, dass Plättchen mit dem unter ihr
liegenden Bindegewebe (Basalmembran, Kollagenfasern etc) in Berührung
kommen. Membranassoziierte heparinähnliche Moleküle verstärken die Inaktivierung von Thrombin und anderen Gerinnungsfaktoren; Thrombomodulin auf der Oberfläche von Endothelzellen bindet Thrombin und verwandelt es in ein Antikoagulans, das Protein C aktiviert und dadurch die Gerinnungsfaktoren Va und VIIIa wirkungslos macht; | Thrombomodulin (ein Rezeptor auf intakten Endothelzellen) bindet Thrombin |
Protein S ist ein Cofaktor des Protein C, das die Thrombusbildung hemmt; | Aktiviertes Protein C (APC) wirkt proteolytisch, hemmt Va und VIIIa und hemmt so die Thrombusbildung |
das membranale TFPI (Tissue factor pathway inhibitor) der Endothelzellen unterbindet die Faktor VIIa- und Xa- Aktivität.  Abbildung: Gerinnungsfördernde (prothrombotsche) Aktivitäten des Endothels Prothrombotisch
( Abbildung): Während ungestörtes Endothel alles tut, um
Gerinnungsvorgänge zu vermeiden (und Thromben abzubauen), ändert sich
das Bild bei Entzündungen oder Verletzungen.
Abbildung: Gerinnungsfördernde (prothrombotsche) Aktivitäten des Endothels Prothrombotisch
( Abbildung): Während ungestörtes Endothel alles tut, um
Gerinnungsvorgänge zu vermeiden (und Thromben abzubauen), ändert sich
das Bild bei Entzündungen oder Verletzungen. Abbildung):
Abbildung): 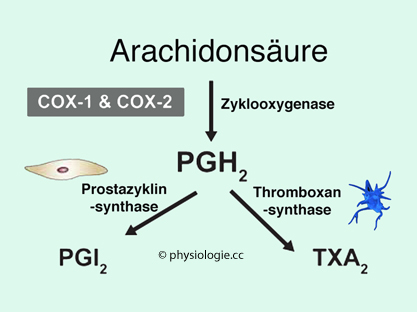 Abbildung: Bildung von Prostacyclin und Thromboxan aus Arachidonsäure / Prostaglandin G
Abbildung: Bildung von Prostacyclin und Thromboxan aus Arachidonsäure / Prostaglandin G
 Acetylsalicylsäure
hemmt die Cyclooxygenase (COX) und damit die Bildung sowohl von
Prostacyclin als auch Thromboxan. Nachdem Endothelzellen innerhalb von
Stunden neue COX (und damit Prostacyclin) bilden können, Thrombozyten
aber keine DNA als Vorlage haben und damit erst neu gebildete Plättchen
wieder Thromboxan synthetisieren, ist die Gerinnungsaktivität des
Blutes durch Acetylsalicylsäure (Aspirin®) für mehrere Tage herabgesetzt. Plättchenkomponente Gerinnungskomponente
Acetylsalicylsäure
hemmt die Cyclooxygenase (COX) und damit die Bildung sowohl von
Prostacyclin als auch Thromboxan. Nachdem Endothelzellen innerhalb von
Stunden neue COX (und damit Prostacyclin) bilden können, Thrombozyten
aber keine DNA als Vorlage haben und damit erst neu gebildete Plättchen
wieder Thromboxan synthetisieren, ist die Gerinnungsaktivität des
Blutes durch Acetylsalicylsäure (Aspirin®) für mehrere Tage herabgesetzt. Plättchenkomponente Gerinnungskomponente An der Oberfläche verletzter Zellen wird Gewebefaktor (TF, tissue factor)
frei. Dieser aktiviert Faktor VII, der wiederum die Faktoren IX und X
aktiviert. Thrombozyten beschleunigen den Vorgang mittels ihrer
Phospholipidmembran. Aktiviertes Thrombin (IIa) fördert die Plättchenaggregation und aktiviert Fibrinogen zu Fibrin (Ia), weiteren Faktor X, sowie Faktor XIII, der das Fibringerinnsel stabilisiert. Abbildung) ist das Beenden von
Blutverlust aus verletzten Gefäßen.
An der Oberfläche verletzter Zellen wird Gewebefaktor (TF, tissue factor)
frei. Dieser aktiviert Faktor VII, der wiederum die Faktoren IX und X
aktiviert. Thrombozyten beschleunigen den Vorgang mittels ihrer
Phospholipidmembran. Aktiviertes Thrombin (IIa) fördert die Plättchenaggregation und aktiviert Fibrinogen zu Fibrin (Ia), weiteren Faktor X, sowie Faktor XIII, der das Fibringerinnsel stabilisiert. Abbildung) ist das Beenden von
Blutverlust aus verletzten Gefäßen.  Abbildung: Aktivierung und Ablauf der Blutstillung an einem verletzten Blutgefäß
Sekunden: Zusammenziehung verletzter Gefäße (Vasokonstriktion), Aneinanderlagerung (Aggregation) und Verschmelzung von Thrombozyten: Primäre Hämostase Minuten: Bildung eines Netzes aus Fibrinfäden durch die Blutgerinnung: Sekundäre Hämostase Tage bis Wochen: Rekonstruktion und Wundheilung. Das Blutgerinnsel (coagulum) wird durch
Fibrinolyse
wieder aufgelöst, sobald es nicht mehr zur Abdichtung benötigt wird. Unter der Wirkung
verschiedener Zytokine
und Kontaktmechanismen wachsen Zellen vor, decken die Wunde
ab und
organisieren das Gewebe neu.Abbildung). Ihr Volumen beträgt ungefähr 10 Femtoliter (Vergleich: Erythrozyten ~90 fl, Lymphozyten ~200 fl). Plättchen und Immunsystem im Knochenmark ( Abbildung).
Abbildung: Aktivierung und Ablauf der Blutstillung an einem verletzten Blutgefäß
Sekunden: Zusammenziehung verletzter Gefäße (Vasokonstriktion), Aneinanderlagerung (Aggregation) und Verschmelzung von Thrombozyten: Primäre Hämostase Minuten: Bildung eines Netzes aus Fibrinfäden durch die Blutgerinnung: Sekundäre Hämostase Tage bis Wochen: Rekonstruktion und Wundheilung. Das Blutgerinnsel (coagulum) wird durch
Fibrinolyse
wieder aufgelöst, sobald es nicht mehr zur Abdichtung benötigt wird. Unter der Wirkung
verschiedener Zytokine
und Kontaktmechanismen wachsen Zellen vor, decken die Wunde
ab und
organisieren das Gewebe neu.Abbildung). Ihr Volumen beträgt ungefähr 10 Femtoliter (Vergleich: Erythrozyten ~90 fl, Lymphozyten ~200 fl). Plättchen und Immunsystem im Knochenmark ( Abbildung). Abbildung: Entstehung von Thrombozyten Thrombopoetin (thrombopoietin THPO, megakaryocyte growth and development factor MGDF)
ist
ein Glykoproteinhormon, das die Megakaryozytopoese - Produktion und
Differenzierung von Blutplättchen - anregt. Es wird vor allem in Leber
(Hepatozyten, Endothelzellen; angeregt durch IL-6) und Niere (proximale Tubuluszellen)
gebildet, ein wenig auch in Knochenmark und quergestreifter Muskulatur.
Abbildung: Entstehung von Thrombozyten Thrombopoetin (thrombopoietin THPO, megakaryocyte growth and development factor MGDF)
ist
ein Glykoproteinhormon, das die Megakaryozytopoese - Produktion und
Differenzierung von Blutplättchen - anregt. Es wird vor allem in Leber
(Hepatozyten, Endothelzellen; angeregt durch IL-6) und Niere (proximale Tubuluszellen)
gebildet, ein wenig auch in Knochenmark und quergestreifter Muskulatur. 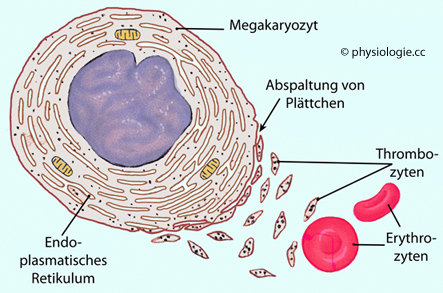 Abbildung: Abschnürung von Thrombozyten aus einem Megakaryozyten
Abbildung: Abschnürung von Thrombozyten aus einem Megakaryozyten Niedrige Plättchenzahl → mehr freies Thrombopoetin → stärkere Einwirkung des Hormons auf Megakaryozyten, verstärkte Plättchenabschnürung Hohe Plättchenzahl → weniger freies Thrombopoetin → geringere Wirkung am Megakaryozytenpool, geringere Thrombozytenbildung
Niedrige Plättchenzahl → mehr freies Thrombopoetin → stärkere Einwirkung des Hormons auf Megakaryozyten, verstärkte Plättchenabschnürung Hohe Plättchenzahl → weniger freies Thrombopoetin → geringere Wirkung am Megakaryozytenpool, geringere Thrombozytenbildung| Bildungsorte des Thrombopoetins sind Leber (hauptsächlich) und Nierenrinde Thrombozyten sind kernlos, verfügen aber über Mitochondrien und mitochondriale Enzyme |
Abbildung)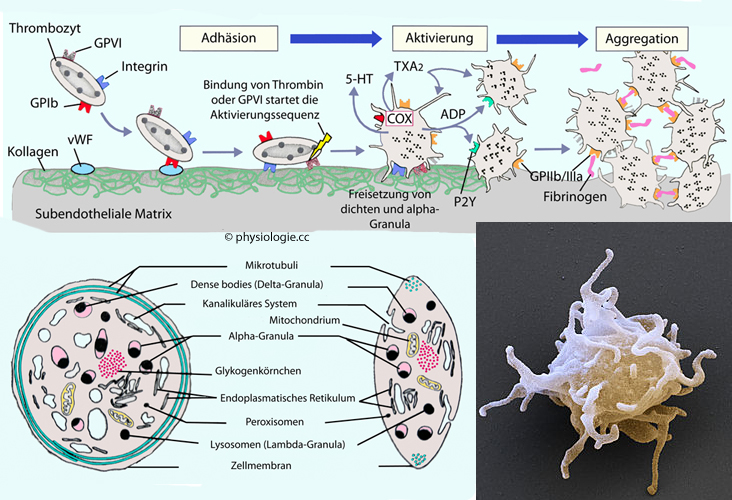 Abbildung: Adhäsion und Aggregation von Thrombozyten an verletzter Gefäßwand (primäre Hämostase)
Abbildung: Adhäsion und Aggregation von Thrombozyten an verletzter Gefäßwand (primäre Hämostase) (vWF)
in
Berührung. Das - im Blutplasma vorhandene - Glykoprotein von Willebrand-Faktor wird von Endothelzellen (hier in Granula, sogenannten Weibel-Palade-Körperchen, gespeichert) und Megakaryozyten gebildet und in Thrombozyten (α-Granula) gespeichert.
(vWF)
in
Berührung. Das - im Blutplasma vorhandene - Glykoprotein von Willebrand-Faktor wird von Endothelzellen (hier in Granula, sogenannten Weibel-Palade-Körperchen, gespeichert) und Megakaryozyten gebildet und in Thrombozyten (α-Granula) gespeichert. | Ist das Endothel verletzt, bindet der vWF einerseits an Kollagenfasern, andererseits an Thrombozyten (über GP-Rezeptoren) |
verformen
sich: Shape change: Pseudopodienbildung ( Abbildung - Lamellopodien, Filopodien - hoher Aktingehalt), sezernieren innerhalb von Sekunden ihren Inhalt (release reaction) - s. Tabelle - und aggregieren (intensiv
gefördert durch ADP), wobei sie Pseudopodien ausbilden, gegenseitig
verschmelzen und einen blutstillenden Plättchenthrombus bilden.  Aktivierte Thrombozyten geben den Großteil ihres Inhalts an
das Serum ab (dadurch steigen hier - im Vergleich zu Blutplasma -
zahlreiche Konzentrationswerte leicht an: Kalium, Phosphat; Serotonin,
Dopamin; LDH, saure Phosphatase u.a.).
Aktivierte Thrombozyten geben den Großteil ihres Inhalts an
das Serum ab (dadurch steigen hier - im Vergleich zu Blutplasma -
zahlreiche Konzentrationswerte leicht an: Kalium, Phosphat; Serotonin,
Dopamin; LDH, saure Phosphatase u.a.).| Inhaltsstoffe von Plättchengranula (Beispiele) |
||
| Substanz |
Granula |
Funktion |
| PDGF, TGFß, FGF |
α |
vasokonstriktorisch mitogen |
| Fibrinogen |
α |
Aggregation über GPIIb/IIIa Gerinnung |
| Fibronektin |
α |
Zelladhäsion |
| vWF |
α |
Adhäsion an Kollagen |
| Gerinnungsfaktoren |
α |
Gerinnung |
| ADP, ATP |
δ |
Aggregation |
| Ca++ |
δ |
Cofaktor |
| Serotonin |
δ |
Plättchenaktivierung Vasokonstriktion |
 α-Granula tragen das Adhäsionsmolekül P-Selektin
in ihrer Membran und enthalten Gerinnungsfaktoren (Fibrinogen,
Fibronektin, Faktor V, Faktor XIII), vWF, Plättchenfaktor 4 (ein
heparinbindendes Chemokin), PDGF, TGF-ß δ-Granula (dense bodies / storage granules) enthalten Ca++-Ionen, ATP / ADP, Serotonin
α-Granula tragen das Adhäsionsmolekül P-Selektin
in ihrer Membran und enthalten Gerinnungsfaktoren (Fibrinogen,
Fibronektin, Faktor V, Faktor XIII), vWF, Plättchenfaktor 4 (ein
heparinbindendes Chemokin), PDGF, TGF-ß δ-Granula (dense bodies / storage granules) enthalten Ca++-Ionen, ATP / ADP, Serotonin| Der GP-IIb/IIIa-Rezeptorkomplex hilft bei der wechselseitigen Verknüpfung von Thrombozyten |
PAF (platelet activating factor)
ist ein Phospholipidaktivator, der mehrere Leukozytenfunktionen - wie
auch Plättchenaggregation und -degranulierung - vermittelt. Er
beteiligt sich an Entzündungsvorgängen
(Chemotaxis, Steigerung der Gefäßpermeabilität). Zahlreiche Zellarten
bilden PAF (Thrombozyten, Endothelzellen, Monozyten / Makrophagen,
neutrophile Granulozyten) - im "Ruhezustand" in geringer Menge, die
Synthese wird bei entsprechender Reizung hinaufreguliert.| ADP regt die Plättchenaggregation an |
Acetylsalicylsäure (Aspirin) hemmt die Thromboxansynthese, was die Gerinnungsaktivität senkt.| Thromboxan A2 wirkt stark vasokonstriktorisch |
| Sinkt die Thromboxansynthese, steigt die Blutungszeit (INR und PTT sind nicht betroffen) |
| Prostazyklin (PGI2) wirkt aggregationshemmend auf Thrombozyten |
| Thrombozytopenie verlängert die Blutungszeit; INR, PTT und Quickwert sind normal |
 Thrombozyten im Vollblut
Thrombozyten im Vollblut Abbildung: Adhäsion und Aggregation von ThrombozytenAbbildung).
Abbildung: Adhäsion und Aggregation von ThrombozytenAbbildung). 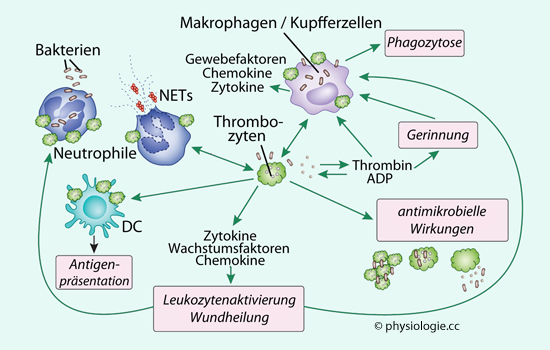 Abbildung: Thrombozyten und Leukozyten kooperieren in der angeborenen Abwehr Verschiedene Pharmaka, wie Aspirin (Acetylsalizylsäure), schwächen die Plättchenfunktion;
Aspirin kann Gerinnselbildungen in den Gefäßen vorbeugen, auf
nüchternen Magen genommen allerdings auch Blutungen der
Magenschleimhaut auslösen.
Abbildung: Thrombozyten und Leukozyten kooperieren in der angeborenen Abwehr Verschiedene Pharmaka, wie Aspirin (Acetylsalizylsäure), schwächen die Plättchenfunktion;
Aspirin kann Gerinnselbildungen in den Gefäßen vorbeugen, auf
nüchternen Magen genommen allerdings auch Blutungen der
Magenschleimhaut auslösen.  Fließfähigkeit einerseits. Diese Qualität (rasche Strömung bei
möglichst geringer Viskosität bis in die Kapillaren) ist - im
"Normalbetrieb" - Voraussetzung für die klaglose Versorgung der Organe
und Gewebe mit Sauerstoff, Nahrungsstoffen, Hormonen, Komponenten des
Immunsystems etc.
Gerinnungsfähigkeit andererseits. Im Falle von Verletzungen der
Gefäßwände muss der Austritt von Blut möglichst rasch und effizient
gestoppt werden. Die Evolution hat dazu ein komplexes System
entwickelt, das garantieren soll, dass Blutungen durch lokale
Viskositätserhöhung zum Stillstand kommen, bevor der Blutverlust die
Funktionsfähigkeit des Kreislaufs beeinträchtigt. Dazu hat sich
offenbar eine mehrgliedrige Kontrolle durch ein komplexes System sich
teils gegenseitig regulierender zellulärer Vorgänge und extrazellulärer Proteine ("Gerinnungsfaktoren") bewährt. aus dem sich kontrahierenden Gerinnsel aus. Vollblut (whole blood) ist die Summe aus Blutzellen und Blutplasma. Blutserum
ist der partikelfreie Überstand des Vollblutes nach vollständiger
Gerinnung. Es ist das in der Labormedizin am meisten verwendete
Untersuchungsmaterial; spektrophotometrische Untersuchungen gelingen
mit ihm störungsfreier als mit Blutplasma (das gerinnen kann). Serum wird gewonnen, indem man geronnenes Blut (mit retrahiertem Blutkuchen) zentrifugiert (10 Minuten bei 2000 x g).
Der Blutkuchen enthält alle Gerinnungsfaktoren (der Proteingehalt des
Serums ist daher um 2-3 g/l geringer als der in Blutplasma - rund 70
g/l).
Fließfähigkeit einerseits. Diese Qualität (rasche Strömung bei
möglichst geringer Viskosität bis in die Kapillaren) ist - im
"Normalbetrieb" - Voraussetzung für die klaglose Versorgung der Organe
und Gewebe mit Sauerstoff, Nahrungsstoffen, Hormonen, Komponenten des
Immunsystems etc.
Gerinnungsfähigkeit andererseits. Im Falle von Verletzungen der
Gefäßwände muss der Austritt von Blut möglichst rasch und effizient
gestoppt werden. Die Evolution hat dazu ein komplexes System
entwickelt, das garantieren soll, dass Blutungen durch lokale
Viskositätserhöhung zum Stillstand kommen, bevor der Blutverlust die
Funktionsfähigkeit des Kreislaufs beeinträchtigt. Dazu hat sich
offenbar eine mehrgliedrige Kontrolle durch ein komplexes System sich
teils gegenseitig regulierender zellulärer Vorgänge und extrazellulärer Proteine ("Gerinnungsfaktoren") bewährt. aus dem sich kontrahierenden Gerinnsel aus. Vollblut (whole blood) ist die Summe aus Blutzellen und Blutplasma. Blutserum
ist der partikelfreie Überstand des Vollblutes nach vollständiger
Gerinnung. Es ist das in der Labormedizin am meisten verwendete
Untersuchungsmaterial; spektrophotometrische Untersuchungen gelingen
mit ihm störungsfreier als mit Blutplasma (das gerinnen kann). Serum wird gewonnen, indem man geronnenes Blut (mit retrahiertem Blutkuchen) zentrifugiert (10 Minuten bei 2000 x g).
Der Blutkuchen enthält alle Gerinnungsfaktoren (der Proteingehalt des
Serums ist daher um 2-3 g/l geringer als der in Blutplasma - rund 70
g/l).| Die Proteinkonzentration von Serum ist geringer (~3%) als in der entsprechenden Blutplasmaprobe |
 Die vollständige Gerinnung von 1 ml Blut erfordert eine mindestens so
große Menge an Faktoren, wie in dieser Menge Blut enthalten ist (2 µg F
VIII, 20 µg
F X, 150 µg Prothrombin, ~2,5 mg Fibrinogen).
Die vollständige Gerinnung von 1 ml Blut erfordert eine mindestens so
große Menge an Faktoren, wie in dieser Menge Blut enthalten ist (2 µg F
VIII, 20 µg
F X, 150 µg Prothrombin, ~2,5 mg Fibrinogen).| Prokoagulatorische Faktoren (F = Faktor, CF = Cofaktor) Nach Boron / Boulpaep: Concise Medical Physiology, Elsevier 2021 |
||
| Name |
alternative Bezeichnung |
Eigenschaften |
| F I |
Fibrinogen |
Globulin |
| F I a |
Fibrin |
|
| F II |
Prothrombin |
Globulin Synthese (Leber) Vit.K-abhängig |
| F II a |
Thrombin |
Serinprotease |
| F III (CF) |
Thromboplastin tissue factor |
integrales Membran-Glykoprotein Rezeptor für F.VIIa aktiv nur in Phospholipidmembran |
| F IV |
Ca++ |
|
| F V |
Proaccelerin |
in Plättchen gespeichert |
| F V a (CF) |
durch ein Ca++-Ion verbundenes Heterodimer |
|
| F VII |
Proconvertin |
Synthese (Leber) Vit.K-abhängig |
| F VII a |
Serinprotease |
|
| F VIII |
Antihämophiler Faktor |
Phospholipidbindende Domäne |
| F VIII a (CF) |
stark homolog zu F V a |
|
| F IX |
Christmas-Faktor PTC |
Synthese (Leber) Vit.K-abhängig |
| F IX a |
Heterodimer (Disulfidbrücke) |
|
| F X |
Stuart-Faktor |
Synthese (Leber) Vit.K-abhängig |
| F X a |
Protease |
|
| F XI |
PTA |
Megakaryozyten synthetisieren, Plättchen speichern es |
| F XI a |
Heterodimer (Disulfidbrücke) | |
| F XII |
Hageman-Faktor |
Glykoprotein (geringere Bedeutung, Mangel führt nicht zu Blutungen) |
| F XII a |
Protease |
|
| F XIII |
Fibrinstabilisierender Faktor (FSF) |
in Plättchen gespeichertes Plasmaprotein |
| F XIII a |
Transglutaminase |
|
| High molecular weight Kininogen |
Fitzgerald-Faktor HMWK |
in Plättchen gespeichertes Plasmaprotein |
| Plasma Präkallikrein |
Fletcher-Faktor |
Plasmaprotein |
| Plasma Kallikrein |
Serinprotease spaltet von HMWK Kallikrein ab |
|
| von Willebrand Faktor |
vWF |
stabilisiert F VIII a fördert Plättchenadhäsion und -aggregation |
Abbildung zeigt ein Gerinnungsschema, das die Beteiligung zellulärer Elemente am Gerinnungsgeschehen betont: Abbildung: Blutgerinnung Abbildung) gelangt an Defektstellen des Gefäßendothels zunächst Gerinnungsfaktor VII (coagulation factor VII, Proconvertin), eine von Hepatozyten Vitamin-K-abhängig produzierte Serinprotease, aus dem Blutplasma an Zellen, die TF (Gewebefaktor, tissue factor, Thromboplastin)
in ihrer Zellmembran haben (und normalerweise nicht für Blutkomponenten
zugäglich sind). Die beiden bilden einen Komplex (TF/VIIa), der Gerinnungsfaktor X (Stuart factor) aktiviert.
Abbildung: Blutgerinnung Abbildung) gelangt an Defektstellen des Gefäßendothels zunächst Gerinnungsfaktor VII (coagulation factor VII, Proconvertin), eine von Hepatozyten Vitamin-K-abhängig produzierte Serinprotease, aus dem Blutplasma an Zellen, die TF (Gewebefaktor, tissue factor, Thromboplastin)
in ihrer Zellmembran haben (und normalerweise nicht für Blutkomponenten
zugäglich sind). Die beiden bilden einen Komplex (TF/VIIa), der Gerinnungsfaktor X (Stuart factor) aktiviert.| Prothrombin (Faktor II) wird durch den Faktor Xa aktiviert (zusammen mit Faktor Va, Ca++ und Phospholipiden) |
 "Bergab": Proteolyse von Fibrinogen, Aktivierung des fibrinstabilisierenden Faktors XIII (FSF) "Bergauf": Aktivierung der Faktoren II (Thrombin aktiviert seine eigene
Vorstufe. ), V und VIII Parakrine Wirkungen: Anregung von Endothelzellen zur Freisetzung von Prostazyklin, NO, ADP, vWF, Plasminaktivator; und von Thrombozyten über PAR-1.Thrombin
aktiviert nicht nur Fibrinogen und führt zur Bildung eines
Fibrinpolymers (Blutgerinnung), es aktiviert auch die Fibrinolyse, also
den Abbau des Fibrinpolymers zu Fibrinfragmenten (Fibrinolyse). Abbildung):
"Bergab": Proteolyse von Fibrinogen, Aktivierung des fibrinstabilisierenden Faktors XIII (FSF) "Bergauf": Aktivierung der Faktoren II (Thrombin aktiviert seine eigene
Vorstufe. ), V und VIII Parakrine Wirkungen: Anregung von Endothelzellen zur Freisetzung von Prostazyklin, NO, ADP, vWF, Plasminaktivator; und von Thrombozyten über PAR-1.Thrombin
aktiviert nicht nur Fibrinogen und führt zur Bildung eines
Fibrinpolymers (Blutgerinnung), es aktiviert auch die Fibrinolyse, also
den Abbau des Fibrinpolymers zu Fibrinfragmenten (Fibrinolyse). Abbildung): Abbildung: Wirkungen aktivierten Thrombins In-vitro Gerinnungshemmung: Durch Zugabe von Komplexbildnern - wie Citrat, Oxalat, EDTA - können Blutproben (z.B. Blutkonserven) ungerinnbar gemacht werden, indem Ca++-Ionen gebunden werden und damit für den Gerinnungsablauf nicht mehr verfügbar sind. s. unten), dadurch deren
Wiksamkeit um 3 Zehnerpotenzen steigert und den Gerinnungsmechanismus
blockiert - sowohl in vitro als auch in vivo.
Abbildung: Wirkungen aktivierten Thrombins In-vitro Gerinnungshemmung: Durch Zugabe von Komplexbildnern - wie Citrat, Oxalat, EDTA - können Blutproben (z.B. Blutkonserven) ungerinnbar gemacht werden, indem Ca++-Ionen gebunden werden und damit für den Gerinnungsablauf nicht mehr verfügbar sind. s. unten), dadurch deren
Wiksamkeit um 3 Zehnerpotenzen steigert und den Gerinnungsmechanismus
blockiert - sowohl in vitro als auch in vivo. | Basophile Granulozyten enthalten Heparin |
Es
bietet eine katalytische Oberfläche an der Verletzungsstelle; es
interagiert spezifisch mit Gerinnungskomponenten; es beschleunigt und
organisiert den Gerinnungsvorgang.| Vitamin K ist erforderlich für die Carboxylierung der Gerinnungsfaktoren II, VII, IX und X ("Prothrombinkomplex") und damit Überführung in die aktive Form (auch für Protein C und S) |
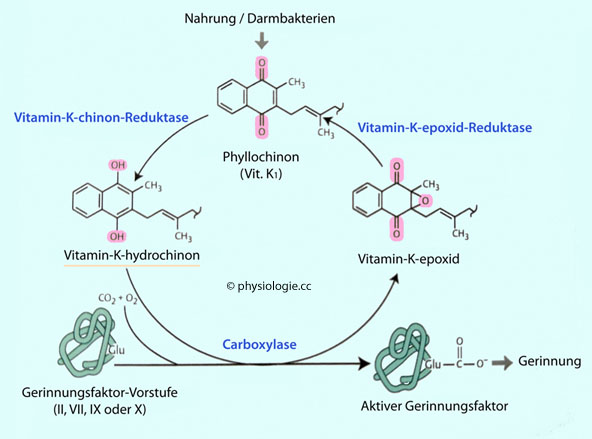
 Abbildung: Vitamine K und was sie für die Carboxylierung von Gerinnungsfaktoren bedeuten Vitamin-K-Antagonisten wie
Phenprocoumon (Marcoumar® - Cumarine) antagonisieren kompetitiv Vit.K in oraler
Dosierung von ~1 mg/d. Sie werden in Tablettenform verabreicht und zur längerfristigen Gerinnungshemmung verwendet. Ihre Wirkung setzt erst mit einer Verzögerung von einigen Tagen
voll ein, da sie die Carboxylierung der neu in der Leber gebildeten
Faktoren II, VII, IX und X blockieren (die bereits im Blut befindlichen
carboxylierten Faktoren müssen erst aus dem Kreislauf entfernt werden).
Endothelzellen sezernieren gerinnungshemmende Proteasen (Gerinnungshemmer), Prostazyklin, NO, Adenosin, sowie den Tissue Factor Pathway Inhibitor (TFPI), ein Plasmaprotein, das an Gerinnungskomplexe (Faktor III + VIIa + Ca++) bindet und die Proteasewirkung von VIIa hemmt. Antithrombin III (AT III) bildet mit mehreren aktivierten Gerinnungsfaktoren stabile Komplexe (IIa, IXa, Xa, XIa, XIIa) und hemmt deren Serinprotease-Aktivität, es baut Thrombin (F IIa) ab und aktiviert an Endothelzellen die Synthese von Plasminogenaktivator (tPA). und Heparansulfat verstärken
die Bindung des AT III an Xa oder Thrombin und damit seine Wirksamkeit (AT III inaktiviert
Thrombin ziemlich langsam, Heparin beschleunigt diese Reaktion um einen
Faktor 103).
Die Gerinnung wird gehemmt. Heparine sind Glykosaminoglykane, die hauptsächlich in Mastzellen und basophilen Granulozyten gebildet und von diesen freigesetzt werden.
Sie finden sich auf der Oberfläche der meisten Zellen, insbesondere auch Endothelzellen. Heparine verringern die Entstehung von Thromben (antithrombotische Wirkung), indem sie die Wirkung des aus der Leber stammenden Plasma-Glykoproteins Antithrombin - ein Protease-Inhibitor, der u.a. Faktoren des intrinsischen (IIa, Xa, XIa, XIIa) und extrinsischen Gerinnungssystems (VIIa) hemmt - über mehrere Mechanismen enorm verstärken.
Abbildung: Vitamine K und was sie für die Carboxylierung von Gerinnungsfaktoren bedeuten Vitamin-K-Antagonisten wie
Phenprocoumon (Marcoumar® - Cumarine) antagonisieren kompetitiv Vit.K in oraler
Dosierung von ~1 mg/d. Sie werden in Tablettenform verabreicht und zur längerfristigen Gerinnungshemmung verwendet. Ihre Wirkung setzt erst mit einer Verzögerung von einigen Tagen
voll ein, da sie die Carboxylierung der neu in der Leber gebildeten
Faktoren II, VII, IX und X blockieren (die bereits im Blut befindlichen
carboxylierten Faktoren müssen erst aus dem Kreislauf entfernt werden).
Endothelzellen sezernieren gerinnungshemmende Proteasen (Gerinnungshemmer), Prostazyklin, NO, Adenosin, sowie den Tissue Factor Pathway Inhibitor (TFPI), ein Plasmaprotein, das an Gerinnungskomplexe (Faktor III + VIIa + Ca++) bindet und die Proteasewirkung von VIIa hemmt. Antithrombin III (AT III) bildet mit mehreren aktivierten Gerinnungsfaktoren stabile Komplexe (IIa, IXa, Xa, XIa, XIIa) und hemmt deren Serinprotease-Aktivität, es baut Thrombin (F IIa) ab und aktiviert an Endothelzellen die Synthese von Plasminogenaktivator (tPA). und Heparansulfat verstärken
die Bindung des AT III an Xa oder Thrombin und damit seine Wirksamkeit (AT III inaktiviert
Thrombin ziemlich langsam, Heparin beschleunigt diese Reaktion um einen
Faktor 103).
Die Gerinnung wird gehemmt. Heparine sind Glykosaminoglykane, die hauptsächlich in Mastzellen und basophilen Granulozyten gebildet und von diesen freigesetzt werden.
Sie finden sich auf der Oberfläche der meisten Zellen, insbesondere auch Endothelzellen. Heparine verringern die Entstehung von Thromben (antithrombotische Wirkung), indem sie die Wirkung des aus der Leber stammenden Plasma-Glykoproteins Antithrombin - ein Protease-Inhibitor, der u.a. Faktoren des intrinsischen (IIa, Xa, XIa, XIIa) und extrinsischen Gerinnungssystems (VIIa) hemmt - über mehrere Mechanismen enorm verstärken.| Heparine verstärken die Wirkung von Antithrombin III und hemmen so die Bildung von Thromben |
Endothelzellen tragen auf ihrer Oberfläche Glykoproteine; diese erschweren die Anheftung von Thrombozyten sowie die Aktivierung kontaktsensibler Faktoren. Thrombomodulin
ist ein endotheliales Glykosaminoglykan, das einen Komplex mit Thrombin
bildet und so die Gerinnung hemmt. Es bindet auch an Protein C: Protein C, in der Leber als Vorstufe Vitamin-K-abhängig gebildet, ist eine gerinnungshemmend, fibrinolytisch und entzündungshemmend wirkende Serinprotease. Zusammen mit
dem Protein S bildet es ein System, das die Bildung von Thrombin
kontrolliert. Das auf
intaktem Endothel immer vorhandene Thrombomodulin sowie der
endotheliale Protein C-Rezeptor (EPCR) aktivieren Protein C. Abbildung) und wirkt dort als Protease,
welche aktives V und VIII lahmlegt (und so die Gerinnung bremst). Der endotheliale Protein C-Rezeptor verstärkt die Aktivierung des Protein C.| Aktiviertes Protein C hemmt die Thrombusbildung |
Protein S, das am Beginn einer Fibrinolyse benötigt wird, wirkt als Cofaktor für Protein C und ist somit ebenfalls ein Antikoagulans. Kupfferzellen der Leber entfernen aktivierte Gerinnungsfaktoren aus der Blutbahn.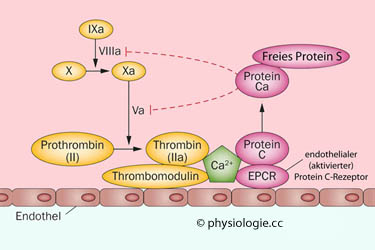 Abbildung: Protein C und S
Abbildung: Protein C und S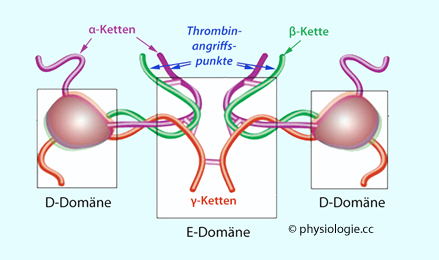 Abbildung: Fibrinogenmolekül Abbildung), wobei
E-Einheiten mit D-Einheiten benachbarter Fibrinmoleküle verknüpft sind.
Dieser Prozess geht mit einer Retraktion des Fibringerinnsels einher
und wird durch den fibrinstabilisierenden Faktor XIII (Laki-Lorand-Faktor) katalysiert.
Abbildung: Fibrinogenmolekül Abbildung), wobei
E-Einheiten mit D-Einheiten benachbarter Fibrinmoleküle verknüpft sind.
Dieser Prozess geht mit einer Retraktion des Fibringerinnsels einher
und wird durch den fibrinstabilisierenden Faktor XIII (Laki-Lorand-Faktor) katalysiert.  Abbildung: Fibrin-Polymerisierung Abbildung).
Das dabei entstandene Fibronmonomer neigt zu spontaner Polymerisierung,
wobei dieses Polymer noch instabil ist und sich leicht wieder auflöst,
also für einen dauerhaften Verschluss ungeeignet ist.
Abbildung: Fibrin-Polymerisierung Abbildung).
Das dabei entstandene Fibronmonomer neigt zu spontaner Polymerisierung,
wobei dieses Polymer noch instabil ist und sich leicht wieder auflöst,
also für einen dauerhaften Verschluss ungeeignet ist.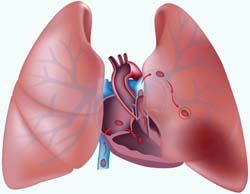 Abbildung: Ein venöser Embolus bleibt im Lungenkreislauf stecken Der Lungenkreislauf hat wichtige Filterfunktion: Thromben ( Abbildung), Luftbläschen
(Injektion), Fettkügelchen (Knochenbruch) bleiben hier stecken und verhindern im Regelfall eine arterielle Embolie. Größere
Thromben können massive parasympathische Reflexe auslösen, die im
Extremfall zu Herzstillstand führen (Lungenembolie).
Abbildung: Ein venöser Embolus bleibt im Lungenkreislauf stecken Der Lungenkreislauf hat wichtige Filterfunktion: Thromben ( Abbildung), Luftbläschen
(Injektion), Fettkügelchen (Knochenbruch) bleiben hier stecken und verhindern im Regelfall eine arterielle Embolie. Größere
Thromben können massive parasympathische Reflexe auslösen, die im
Extremfall zu Herzstillstand führen (Lungenembolie). Abbildung: Thrombozyten-, Gerinnungs- und
Fibrinolysesystem
Abbildung: Thrombozyten-, Gerinnungs- und
Fibrinolysesystem Fi, unlösliches (insoluble) Fibrin Fs, lösliches (soluble) Fibrin H, Heparin
Plasminogenaktivatoren (PA) sind Serinproteasen, die aus Plasminogen proteolytisch Plasmin freisetzen.
Die wichtigsten Formen sind Gewebeaktivator (tPA) und Urokinase (uPA). Die
Aktivität der Plasminogenaktivatoren wird durch PA-Inhibitoren (1 und
2) sowie durch den Protein C-Inhibitor gehemmt. Abbildung), indem es Fibrin in wasserlösliche
Bruchstücke zerlegt (Fibrinolyse). Gleichzeitig baut es auch Fibrinogen und andere
Gerinnungsfaktoren (V, VIII, IX, XI, XII) ab, reduziert also insgesamt
die Gerinnungsfähigkeit des Blutes.
Fi, unlösliches (insoluble) Fibrin Fs, lösliches (soluble) Fibrin H, Heparin
Plasminogenaktivatoren (PA) sind Serinproteasen, die aus Plasminogen proteolytisch Plasmin freisetzen.
Die wichtigsten Formen sind Gewebeaktivator (tPA) und Urokinase (uPA). Die
Aktivität der Plasminogenaktivatoren wird durch PA-Inhibitoren (1 und
2) sowie durch den Protein C-Inhibitor gehemmt. Abbildung), indem es Fibrin in wasserlösliche
Bruchstücke zerlegt (Fibrinolyse). Gleichzeitig baut es auch Fibrinogen und andere
Gerinnungsfaktoren (V, VIII, IX, XI, XII) ab, reduziert also insgesamt
die Gerinnungsfähigkeit des Blutes. Abbildung: Fibrinolytisches SystemDie
Fibrinolyse wird synchron mit der Blutgerinnung aktiviert,
ihr Wirkungseintritt ist aber verzögert, sodass genug Zeit bleibt, im
Fall einer Verletzung die Hämostase ausreichend zur Wirkung kommen zu
lassen. Abbildung). Die Produktion von PAI wird durch Thrombin und einige Zytokine angeregt.
Abbildung: Fibrinolytisches SystemDie
Fibrinolyse wird synchron mit der Blutgerinnung aktiviert,
ihr Wirkungseintritt ist aber verzögert, sodass genug Zeit bleibt, im
Fall einer Verletzung die Hämostase ausreichend zur Wirkung kommen zu
lassen. Abbildung). Die Produktion von PAI wird durch Thrombin und einige Zytokine angeregt. Endothelialer Gewebeaktivator t-PA (tissue plasminogen activator),
der an Fibrin gebunden besonders aktiv wird. Diese Serinprotease wird
von Endothelzellen gebildet und verwandelt Plasminogen zur
fibrinolytisch wirkenden Protease Plasmin
Endothelialer Gewebeaktivator t-PA (tissue plasminogen activator),
der an Fibrin gebunden besonders aktiv wird. Diese Serinprotease wird
von Endothelzellen gebildet und verwandelt Plasminogen zur
fibrinolytisch wirkenden Protease Plasmin| Rekombinanter tPA dient therapeutisch als Thrombolyse-Aktivator |
Streptokinase, ein Produkt von Streptokokken; es bildet einen Komplex mit Plasminogen, der weiteres Plasminogen aktiviert Zur Thrombolyse verwendet man rekombinanten tPA und Streptokinase Kontaktaktivierte Faktoren der endogenen Gerinnungs-Vorphase (XIa, XIIa) Plasminogenaktivatoren aus Fibroblasten, Leukozyten und anderen Zellen Urokinase in den Nierentubuli (Urokinase beugt einer Gerinnung in den Harnwegen vor). | Urokinase aktiviert Plasmin und löst Fibringerinnsel auf |
| α2-Antiplasmin hemmt die Fibrinolyse |
Substanzen wie Streptokinase
aktivieren Plasminogen und lösen Thromben auf; sie werden z.B. zur
Bekämpfung von Schlaganfällen oder Herzinfarkten eingesetzt. In
Thromben findet sich wenig Antiplasmin (es kann nur schwer in sie
eindringen) und das Plasmin kann hier gut wirken - im Gegensatz zum
freien Plasma, wo es ständig durch Antiplasmin abgebaut wird. Katecholamine und Bradykinin steigern den t-PA-Spiegel Serin-Protease-Inhibitoren (Serpine) senken den t-PA-Spiegel: Die Plasminogen-Aktivator-Inhibitoren
PAI-1 (hauptsächlich aus Endothelzellen: Komplexiert mit t-PA) und
PAI-2 (wird von der Plazenta gebildet und könnte das Thromboserisiko
während der Schwangerschaft beeinflussen).| Endogene Faktoren in Fibrinolyse und Antikoagulation Nach Silverthorn, Human Physiology - an integrated approach, 4th ed. Pearson International 2007 |
||||
| Faktor |
Quelle |
Aktiviert oder freigesetzt in Antwort auf |
Rolle bei Gerinnungshemmung oder Fibrinolyse |
Weitere Rollen / Kommentare |
| Plasminogen und Plasmin |
Leber Plasma |
t-PA und Thrombin |
Löst Fibrin und Fibrinogen auf |
-- |
| t-PA |
Viele Gewebe |
Plasmaspiegel steigt mit Stress und Protein C |
Aktiviert Plasminogen |
Rekombiniertes t-PA klinisch für Plättchenauflösung |
| Antithrombin III |
Leber Plasma |
-- |
Antikoagulans Blockiert Faktoren IX, X, XI, XII, Thrombin, Kallikrein |
Verstärkt durch Heparin |
| Prostazyklin |
Endothelzellen |
-- |
Blockiert Plättchenaggregation |
Vasodilatator |
 Testung des Gerinnungssystems (Blutplasma) (Thromboplastinzeit TPZ, Prothrombinzeit): 11-16 Sekunden (Referenzbereich 70-130%)
Testung des Gerinnungssystems (Blutplasma) (Thromboplastinzeit TPZ, Prothrombinzeit): 11-16 Sekunden (Referenzbereich 70-130%)
| Die PTT testet das intrinsische System und ist bei Faktor-VII-Mangel nicht verändert Verlängerte PTT bei sonst normalen Gerinnungswerten legt Faktor VIII-Mangel nahe |
Die Aktivität des extrinsischen Gerinnungssystems wird heute über den INR-Wert
(International Normalized Ratio) quantifiziert: Normalerweise bei 1
(gemessen / normal; Quotient
bezogen auf die normale Prothrombinzeit = "Quick-Wert") gelegen, steigt er (etwa durch Cumarine) infolge
Hemmung der Gerinnungsaktivität (längere Gerinnungszeit), z.B. wird bei
Herzklappenpatienten ein Wert zwischen 2 und 3 angestrebt, um der Bildung von
Thromben verlässlich vorzubeugen. Bei erhöhtem INR steigt allerdings die
Blutungsgefahr, eine sorgfältige Nutzen/Risken-Abwägung
ist angebracht. Abbildung: Thrombelastometer, Schema des Messteils (links unten) und Thrombelastogramm (rechts unten) Abbildung). ), die auch für Plättchenfunktionsstörungen
charakteristisch sind. Verbrauchskoagulopathie
bedeutet, dass es bei disseminierter intravaskulärer Koagulation zu
einem Verbrauch von Gerinnungsfaktoren und dadurch (systemisch) zu
Senkung der Gerinnungsaktivität kommt. Verursacht wird dies z.B. durch
Sepsis, multiple Traumen, oder auch vorzeitige Plazentarablösung:
Thromben treten an verschiedenen Stellen des Körpers auf, es kommt zu
Einschränkungen der Durchblutung, ischämiebedingten Funktionseinbußen
und massiven Gerinnungsstörungen.
Abbildung: Thrombelastometer, Schema des Messteils (links unten) und Thrombelastogramm (rechts unten) Abbildung). ), die auch für Plättchenfunktionsstörungen
charakteristisch sind. Verbrauchskoagulopathie
bedeutet, dass es bei disseminierter intravaskulärer Koagulation zu
einem Verbrauch von Gerinnungsfaktoren und dadurch (systemisch) zu
Senkung der Gerinnungsaktivität kommt. Verursacht wird dies z.B. durch
Sepsis, multiple Traumen, oder auch vorzeitige Plazentarablösung:
Thromben treten an verschiedenen Stellen des Körpers auf, es kommt zu
Einschränkungen der Durchblutung, ischämiebedingten Funktionseinbußen
und massiven Gerinnungsstörungen. | Bei Von-Willebrand-Jürgens-Syndrom (Mangel an Faktor VIII und vWF) sind Blutungszeit und PTT verlängert |
bezeichnet, Gerinnungsstörungen als
Koagulopathien. Gendefekte können die normale Synthese von Gerinnungsfaktoren
verhindern. Bluterkrankheiten bezeichnet man als Hämophilien. Bei der am häufigsten auftretenden Form, der Hämophilie A,
besteht ein Mangel an Faktor VIII; unzureichende Bildung des Faktors IX
bedingt die Hämophilie B.
Eine Thrombose
liegt vor, wenn sich an den Gefäßwänden Zellaggregate
und Gerinnsel ausbilden. Diese engen den Blutstrom ein oder verlegen
das Gefäß. Thrombenbildung wird gefördert durch langsame Blutströmung
(Bettlägrigkeit, Immobilisierung - "economy class syndrome"), erhöhte
Gerinnungsneigung (z.B. gestörte Fibrinolyse), Bluteindickung (z.B.
"Blutdoping") und Gefäßveränderungen (z.B. Atheromatose). Gerinnungshemmung wird klinisch vor allem (akut) mittels Heparin und (über längere Zeit) mit Vitamin-K-Antagonisten (Cumarinderivaten) bewirkt. Auch Hirudin - ein Gemisch von Peptiden im Speichel des Blutegels - kann zum Einsatz kommen. 
 Das System der
Hämostase / Koagulation / Thrombenbildung balanciert pro- und
antikoagulatorische sowie pro- und antifibrinolytische Komponenten. Der Großteil der Vorgänge erfolgt auf der Oberfläche von Endothelzellen und Thrombozyten. Endotheliale Faktoren hemmen das Aneinanderlagern von
Blutplättchen und die Gerinnung und fördern die Lyse von Thromben (gerinnungshemmende Proteasen, Prostazyklin, NO, Adenosin, TFPI). Endothel (von Willebrand-Faktor vWF, Prostazyklin, Plasminogenaktivator t-PA) und Gefäßmuskulatur liefern die Gefäßkomponente, Thrombozyten (Vernetzung, Gerinnungsfaktoren, Ca++) die Plättchenkomponente, Koagulation und Fibrinolyse die Gerinnungskomponente
der Hämostase Hämostase erfolgt binnen Sekunden
(Vasokonstriktion, Plättchenaggregation) bis Minuten (Bildung eines Fibrinnetzes). Thromboxan A2 wirkt vasokonstriktorisch. Bei Endothelschäden verbindet vWF Kollagenfasern mit Thrombozyten.
Nach Kontakt mit verletztem Endothel aggregieren die Plättchen,
freigesetztes Serotonin bindet an plättcheneigene 5-HT2A-Rezeptoren.
Das beschleunigt die Aggregation, der Plättchenthrombus verfestigt
sich (Prostazyklin wirkt aggregationshemmend). Serotonin, Adrenalin, Ca++
aus aktivierten Thrombozyten bewirken lokale Vasokonstriktion, diese
unterstützt
Blutstillung und Defektheilung. Thrombopoetin
(Leber, Niere, Knochenmark) regt die Bildung von Plättchen an; diese werden nach 8-10 Tagen im Kreislauf
vor allem in der Milz abgebaut. Angeregte Bildung
(Blutverlust) oder Entfernung der Milz kann ihre Zahl von 0,15-0,30 auf
~1 Mio/µl steigern. Zur stabilen
Anhaftung von Thrombozyten ist die Aktivierung des
Glykoprotein-IIb/IIIa-Rezeptorkomplexes erforderlich. Thrombozytopenie ist eine verminderte Plättchenzahl, bis
~30 Tausend/µl tritt keine Blutungsneigung auf (funktionelle
Reserve), darunter petechiale Blutungen,
unter 10 Tausend/µl Spontanblutungen. Thrombozytopenie verlängert die Blutungszeit; INR, PTT und Quickwert sind normal. Thrombozyten wirken direkt antimikrobiell, setzen entzündungsfördernde, chemotaktische und
Wachstumsfaktoren frei, was Leukozyten anlockt und aktiviert Die vollständige Gerinnung erfordert eine mindestens so große Menge an Faktoren (meist mit römischen Ziffern bezeichnet, die aktivierte Form mit “a”), wie im betreffenden Volumen Blut enthalten ist. Sie beginnt mit
der Aktivierung der Vorphase, was auf zwei Wegen erfolgen kann - durch
das “intrinsische” und “extrinsische” System. Ersteres beginnt mit der
Aktivierung von Thrombozyten und F.XII (Hageman-Faktor) an
verletzten Oberflächen, dann werden die Faktoren XI, IX und VIII aktiviert.
Faktor VIII wird unterstützt durch den vWF. Das
extrinsische System zündet bei Verletzung durch freigesetzte
Zellstrukturen und Thromboplastin (Gewebefaktor), hier wirkt der Faktor
VII (Prokonvertin). Die Systeme
münden in eine gemeinsame Endstrecke, in welche die Faktoren X
(Stuart-Prower-Faktor) und V (Proaccelerin) eingeschaltet sind. Die
Anwesenheit freier Ca++ ist notwendig (Zugabe von Citrat, Oxalat, EDTA macht Blutproben ungerinnbar). Die Reaktionskaskaden bilden innerhalb weniger Minuten einen Prothrombinaktivator - dieser wandelt Prothrombin (F.II) in Thrombin um. Vitamin K ist erforderlich für die Carboxylierung der Gerinnungsfaktoren II, VII, IX und X ("Prothrombinkomplex"). Fibrinogen (F.I, ~3 g/l)
wird durch Gerinnungs- und Plättchenfaktoren zu Fibrin, dieses bildet
ein Koagulum, das durch F.XIII (Laki-Lorand-Faktor) stabilisiert und durch Retraktin zu
einem widerstandsfähigen Netz wird; dabei tritt Serum aus Zu Gerinnungshemmern zählen Antithrombin III (AT III), es bildet stabile Komplexe mit IIa, IXa, Xa, XIa, XIIa, baut Thrombin (IIa) ab und aktiviert an Endothelzellen die Synthese von Plasminogenaktivator (tPA). Heparine (aus Mastzellen, basophilen Granulozyten) beschleunigen seine Wirksamkeit um den Faktor 1000. Protein C wirkt gerinnungshemmend, fibrinolytisch und entzündungshemmend, es bremst zusammen mit Protein S
die Bildung von Thrombin durch Abbau aktiven F.V und F.VIII. Die Lunge
ist eine wichtige Filterstation, sie fängt Thromben, Fett- und
Luftblasen auf und verhindert ihre Passage in den arteriellen
Kreislauf, wo sie zu Embolisierung führen können Die Fibrinolyse wird synchron mit der Blutgerinnung aktiviert, ihr Wirkungseintritt ist verzögert. Plasmin wird durch zahlreiche Aktivatoren (endothelial, leukozytär, Urokinase) aus Plasminogen freigesetzt
und löst Thromben auf, indem es Fibrin und andere Gerinnungsfaktoren abbaut. Freie
Plasminmoleküle werden im Blut inaktiviert, die fibrinolytische
Aktivität bleibt auf Stellen begrenzt, wo sich ein Thrombus gebildet
hat - sie ist regional unterschiedlich
(endothelabhängig): Balance zwischen koagulatorischen und
antikoagulatorischen Effekten u.a. durch Freisetzung eines
Plasminogenaktivator-Inhibitors (PAI) mit prokoagulatorischer Wirkung. α2-Antiplasmin hemmt die Fibrinolyse Die
Blutungszeit prüft das gesamte Hämostasesystem (2-10 Minuten,
methodenabhängig), die Gerinnungszeit (6-15 Minuten) das gesamte
Gerinnungssystem, der Quick-test (Prothrombinzeit) das extrinsische
(~14 Sekunden), die partielle Thromboplastinzeit (PTT) das intrinsische
System (~30 Sekunden). Das extrinsische Systems wird über den INR-Wert
(International Normalized Ratio: gemessen / normal)
quantifiziert: Normalerweise 1,0. Thrombelastometer registrieren Stärke
und zeitlichen Ablauf der Viskositätsveränderung einer
gerinnungsaktivierten Blutprobe (→Thrombelastogramm).
Gerinnungshemmung erfolgt mittels Heparin (Sofortwirkung) und (über
längere Zeit) Vitamin-K-Antagonisten (Cumarinderivaten) Das System der
Hämostase / Koagulation / Thrombenbildung balanciert pro- und
antikoagulatorische sowie pro- und antifibrinolytische Komponenten. Der Großteil der Vorgänge erfolgt auf der Oberfläche von Endothelzellen und Thrombozyten. Endotheliale Faktoren hemmen das Aneinanderlagern von
Blutplättchen und die Gerinnung und fördern die Lyse von Thromben (gerinnungshemmende Proteasen, Prostazyklin, NO, Adenosin, TFPI). Endothel (von Willebrand-Faktor vWF, Prostazyklin, Plasminogenaktivator t-PA) und Gefäßmuskulatur liefern die Gefäßkomponente, Thrombozyten (Vernetzung, Gerinnungsfaktoren, Ca++) die Plättchenkomponente, Koagulation und Fibrinolyse die Gerinnungskomponente
der Hämostase Hämostase erfolgt binnen Sekunden
(Vasokonstriktion, Plättchenaggregation) bis Minuten (Bildung eines Fibrinnetzes). Thromboxan A2 wirkt vasokonstriktorisch. Bei Endothelschäden verbindet vWF Kollagenfasern mit Thrombozyten.
Nach Kontakt mit verletztem Endothel aggregieren die Plättchen,
freigesetztes Serotonin bindet an plättcheneigene 5-HT2A-Rezeptoren.
Das beschleunigt die Aggregation, der Plättchenthrombus verfestigt
sich (Prostazyklin wirkt aggregationshemmend). Serotonin, Adrenalin, Ca++
aus aktivierten Thrombozyten bewirken lokale Vasokonstriktion, diese
unterstützt
Blutstillung und Defektheilung. Thrombopoetin
(Leber, Niere, Knochenmark) regt die Bildung von Plättchen an; diese werden nach 8-10 Tagen im Kreislauf
vor allem in der Milz abgebaut. Angeregte Bildung
(Blutverlust) oder Entfernung der Milz kann ihre Zahl von 0,15-0,30 auf
~1 Mio/µl steigern. Zur stabilen
Anhaftung von Thrombozyten ist die Aktivierung des
Glykoprotein-IIb/IIIa-Rezeptorkomplexes erforderlich. Thrombozytopenie ist eine verminderte Plättchenzahl, bis
~30 Tausend/µl tritt keine Blutungsneigung auf (funktionelle
Reserve), darunter petechiale Blutungen,
unter 10 Tausend/µl Spontanblutungen. Thrombozytopenie verlängert die Blutungszeit; INR, PTT und Quickwert sind normal. Thrombozyten wirken direkt antimikrobiell, setzen entzündungsfördernde, chemotaktische und
Wachstumsfaktoren frei, was Leukozyten anlockt und aktiviert Die vollständige Gerinnung erfordert eine mindestens so große Menge an Faktoren (meist mit römischen Ziffern bezeichnet, die aktivierte Form mit “a”), wie im betreffenden Volumen Blut enthalten ist. Sie beginnt mit
der Aktivierung der Vorphase, was auf zwei Wegen erfolgen kann - durch
das “intrinsische” und “extrinsische” System. Ersteres beginnt mit der
Aktivierung von Thrombozyten und F.XII (Hageman-Faktor) an
verletzten Oberflächen, dann werden die Faktoren XI, IX und VIII aktiviert.
Faktor VIII wird unterstützt durch den vWF. Das
extrinsische System zündet bei Verletzung durch freigesetzte
Zellstrukturen und Thromboplastin (Gewebefaktor), hier wirkt der Faktor
VII (Prokonvertin). Die Systeme
münden in eine gemeinsame Endstrecke, in welche die Faktoren X
(Stuart-Prower-Faktor) und V (Proaccelerin) eingeschaltet sind. Die
Anwesenheit freier Ca++ ist notwendig (Zugabe von Citrat, Oxalat, EDTA macht Blutproben ungerinnbar). Die Reaktionskaskaden bilden innerhalb weniger Minuten einen Prothrombinaktivator - dieser wandelt Prothrombin (F.II) in Thrombin um. Vitamin K ist erforderlich für die Carboxylierung der Gerinnungsfaktoren II, VII, IX und X ("Prothrombinkomplex"). Fibrinogen (F.I, ~3 g/l)
wird durch Gerinnungs- und Plättchenfaktoren zu Fibrin, dieses bildet
ein Koagulum, das durch F.XIII (Laki-Lorand-Faktor) stabilisiert und durch Retraktin zu
einem widerstandsfähigen Netz wird; dabei tritt Serum aus Zu Gerinnungshemmern zählen Antithrombin III (AT III), es bildet stabile Komplexe mit IIa, IXa, Xa, XIa, XIIa, baut Thrombin (IIa) ab und aktiviert an Endothelzellen die Synthese von Plasminogenaktivator (tPA). Heparine (aus Mastzellen, basophilen Granulozyten) beschleunigen seine Wirksamkeit um den Faktor 1000. Protein C wirkt gerinnungshemmend, fibrinolytisch und entzündungshemmend, es bremst zusammen mit Protein S
die Bildung von Thrombin durch Abbau aktiven F.V und F.VIII. Die Lunge
ist eine wichtige Filterstation, sie fängt Thromben, Fett- und
Luftblasen auf und verhindert ihre Passage in den arteriellen
Kreislauf, wo sie zu Embolisierung führen können Die Fibrinolyse wird synchron mit der Blutgerinnung aktiviert, ihr Wirkungseintritt ist verzögert. Plasmin wird durch zahlreiche Aktivatoren (endothelial, leukozytär, Urokinase) aus Plasminogen freigesetzt
und löst Thromben auf, indem es Fibrin und andere Gerinnungsfaktoren abbaut. Freie
Plasminmoleküle werden im Blut inaktiviert, die fibrinolytische
Aktivität bleibt auf Stellen begrenzt, wo sich ein Thrombus gebildet
hat - sie ist regional unterschiedlich
(endothelabhängig): Balance zwischen koagulatorischen und
antikoagulatorischen Effekten u.a. durch Freisetzung eines
Plasminogenaktivator-Inhibitors (PAI) mit prokoagulatorischer Wirkung. α2-Antiplasmin hemmt die Fibrinolyse Die
Blutungszeit prüft das gesamte Hämostasesystem (2-10 Minuten,
methodenabhängig), die Gerinnungszeit (6-15 Minuten) das gesamte
Gerinnungssystem, der Quick-test (Prothrombinzeit) das extrinsische
(~14 Sekunden), die partielle Thromboplastinzeit (PTT) das intrinsische
System (~30 Sekunden). Das extrinsische Systems wird über den INR-Wert
(International Normalized Ratio: gemessen / normal)
quantifiziert: Normalerweise 1,0. Thrombelastometer registrieren Stärke
und zeitlichen Ablauf der Viskositätsveränderung einer
gerinnungsaktivierten Blutprobe (→Thrombelastogramm).
Gerinnungshemmung erfolgt mittels Heparin (Sofortwirkung) und (über
längere Zeit) Vitamin-K-Antagonisten (Cumarinderivaten) |
