

Eine Reise durch die Physiologie - Wie der Körper des Menschen funktioniert


 Acetylcholin, Amine, Purine, Peptide, lokale Mediatoren
Acetylcholin, Amine, Purine, Peptide, lokale Mediatoren
 Aden(os)in: ἀδήν = Drüse (Namensgebung durch Albrecht Kossel)
Aden(os)in: ἀδήν = Drüse (Namensgebung durch Albrecht Kossel)| Die Informationsübermittlung von Zelle zu Zelle folgt bestimmten Regeln: Spezifität (passende Rezeptoren), Verstärkung (Enzymwirkung, viele second-messenger-Moleküle) und Modifikation der Wirkung (über zusätzliche Signalmoleküle). Extrazelluläre Signalstoffe sind Hormone, Neurotransmitter (wie Acetylcholin und Katecholamine), Zytokine (im Immunsystem), Eikosanoide (Prostaglandine, Leukotriene), Purine (ATP, Adenosin) und Gase (wie Stickstoffmonoxid). Acetylcholin ist der Transmitter aller Neuriten, die das Zentralnervensystem verlassen (an motorischen Endplatten der Skelettmuskulatur, an der Endigung präganglionärer autonomer Fasern). Acetylcholin wirkt - je nach Schaltstelle - über nikotinische (ionotrope) oder muskarinische (G-Protein-gekoppelte) Rezeptoren. Katecholamine kommen im Gehirn (Dopamin, Noradrenalin), als postganglionär-sympathische Transmitter (hauptsächlich Noradrenalin) und als Nebennierenmarkhormon vor (hauptsächlich Adrenalin); sie wirken über D-Rezeptoren (Dopamin) bzw. α- und β-Rezeptoren (Noradrenalin, Adrenalin). Biogene Amine - Serotonin und Histamin - Peptide wie z.B. Endotheline, Neuropeptid Y (NPY), das vasoaktive intestinale Peptid (VIP), Eikosanoide und Gasotransmitter wirken überwiegend über G-Protein-Rezeptoren an zahlreichen Geweben in sehr unterschiedlicher Weise. |
 Acetylcholin Purinerge Transmission Katecholamine Histamin Serotonin Peptide Gasotransmitter Eikosanoide (Prostaglandine, Thromboxane, Leukotriene) Kininsystem
Acetylcholin Purinerge Transmission Katecholamine Histamin Serotonin Peptide Gasotransmitter Eikosanoide (Prostaglandine, Thromboxane, Leukotriene) Kininsystem
 Amine Katecholamine Neuropeptide Proteinkinase B
Amine Katecholamine Neuropeptide Proteinkinase B
 Core messages
Core messages
 Abbildung: Cholinerge, adrenerge und dopaminerge Transmission im vegetativen System Als Mediator bezeichnet man einen Signalstoff, wenn er auf kurze Distanz spezifisch auf Nachbarzellen wirkt (Beispiel Purine). Ein (Neuro-)Transmitter wirkt an synaptischen Strukturen - auf kurze Distanz für begrenzte Zeit. Ein Zytokin ist eine Signalsubstanz des Immunsystems (es kann modulatorisch oder wie ein Hormon wirken). Hormone werden an den Kreislauf abgegeben und können überall im Körper wirksam werden (soferne sie auf aktivierbare Rezeptoren koppeln). Die Übergänge zwischen diesen (willkürlich erstellten) Kategorien sind fließend.
Spezifität (Schlüssel-Schloss-Prinzip) - nur der "richtige" Signalstoff
löst eine Wirkung aus (allerdings können ähnliche Moleküle ebenfalls am
Rezeptor binden, diese regen entweder den Signalweg an - Agonisten - oder sie hemmen ihn - Antagonisten)
Verstärkung - wenn der Signalstoff den betreffenden Rezeptor
"einschaltet", kann es zu vielfacher molekularer Sekundärreaktion
kommen und sich die Wirkung multiplizieren. Das
kann mehrere hintereinandergeschaltete Vorgänge betreffen (second
messenger ... Enzymaktivierung), sodass die Verstärkung mehrere
Zehnerpotenzen ausmachen kann
Zusätzliche Regelung - solche komplexe molekulare
Übersetzungsmechanismen bieten mehrfache Möglichkeiten der
Querbeeinflussung durch (hemmende, fördernde, steuernde)
Zusatzeinflüsse (Kofaktoren, Modifikatoren)
Abbildung: Cholinerge, adrenerge und dopaminerge Transmission im vegetativen System Als Mediator bezeichnet man einen Signalstoff, wenn er auf kurze Distanz spezifisch auf Nachbarzellen wirkt (Beispiel Purine). Ein (Neuro-)Transmitter wirkt an synaptischen Strukturen - auf kurze Distanz für begrenzte Zeit. Ein Zytokin ist eine Signalsubstanz des Immunsystems (es kann modulatorisch oder wie ein Hormon wirken). Hormone werden an den Kreislauf abgegeben und können überall im Körper wirksam werden (soferne sie auf aktivierbare Rezeptoren koppeln). Die Übergänge zwischen diesen (willkürlich erstellten) Kategorien sind fließend.
Spezifität (Schlüssel-Schloss-Prinzip) - nur der "richtige" Signalstoff
löst eine Wirkung aus (allerdings können ähnliche Moleküle ebenfalls am
Rezeptor binden, diese regen entweder den Signalweg an - Agonisten - oder sie hemmen ihn - Antagonisten)
Verstärkung - wenn der Signalstoff den betreffenden Rezeptor
"einschaltet", kann es zu vielfacher molekularer Sekundärreaktion
kommen und sich die Wirkung multiplizieren. Das
kann mehrere hintereinandergeschaltete Vorgänge betreffen (second
messenger ... Enzymaktivierung), sodass die Verstärkung mehrere
Zehnerpotenzen ausmachen kann
Zusätzliche Regelung - solche komplexe molekulare
Übersetzungsmechanismen bieten mehrfache Möglichkeiten der
Querbeeinflussung durch (hemmende, fördernde, steuernde)
Zusatzeinflüsse (Kofaktoren, Modifikatoren) Abbildung):
Abbildung):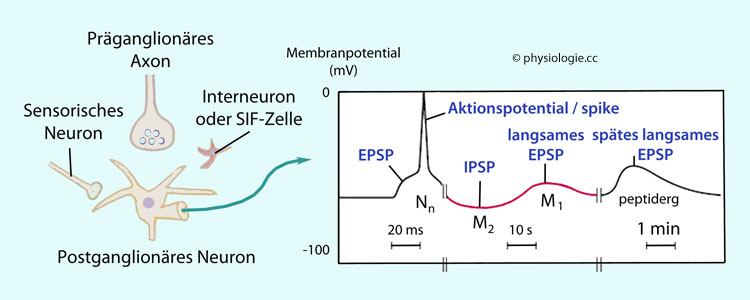 Abbildung: Postsynaptische Potentiale nach präsynaptischer Reizung in einem autonomen Ganglion
Abbildung: Postsynaptische Potentiale nach präsynaptischer Reizung in einem autonomen Ganglion das erste (frühe) EPSP aus der Aktivierung nikotinerger (Nn) Rezeptoren, was zu Na+- (und Ca++-) Einstrom führt; bei Überschwelligkeit tritt ein Aktionspotential (spike) auf.
Es folgen weitere Potentialschwankungen: ein 2-5 Sekunden andauerndes IPSP, hauptsächlich durch Aktivierung
muskarinerger (M2) Rezeptoren, das die Kalium-Leitfähigkeit erhöht (andere Transmitter - wie Dopamin oder Adenosin - sind ebenfalls beteiligt), ein langsames, etwa 10 Sekunden dauerndes EPSPs durch Aktivierung
muskarinerger (M1) Rezeptoren, welche Kaliumkanäle schließen, ein weiteres langsames, spätes EPSPs - das 1-2 Minuten anhalten kann - durch Aktivierung peptiderger Rezeptoren (u.a. Substanz P, GnRH-ähnliche Peptide), was ebenfalls die K+-Leitfähigkeit der Membran herabsetzt.
das erste (frühe) EPSP aus der Aktivierung nikotinerger (Nn) Rezeptoren, was zu Na+- (und Ca++-) Einstrom führt; bei Überschwelligkeit tritt ein Aktionspotential (spike) auf.
Es folgen weitere Potentialschwankungen: ein 2-5 Sekunden andauerndes IPSP, hauptsächlich durch Aktivierung
muskarinerger (M2) Rezeptoren, das die Kalium-Leitfähigkeit erhöht (andere Transmitter - wie Dopamin oder Adenosin - sind ebenfalls beteiligt), ein langsames, etwa 10 Sekunden dauerndes EPSPs durch Aktivierung
muskarinerger (M1) Rezeptoren, welche Kaliumkanäle schließen, ein weiteres langsames, spätes EPSPs - das 1-2 Minuten anhalten kann - durch Aktivierung peptiderger Rezeptoren (u.a. Substanz P, GnRH-ähnliche Peptide), was ebenfalls die K+-Leitfähigkeit der Membran herabsetzt. vgl. dort
vgl. dort

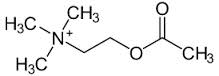 Acetylcholin
Acetylcholin Der österreichische Pharmakologe Otto Loewi
konnte 1921 experimentell nachweisen, dass die parasympathische Wirkung
auf das Herz durch einen "Vagusstoff" vermittelt wird. Dieser wurde vom britischen Pharmakologen Sir Henry Dale als Acetylcholin identifiziert (eine
Rolle des Acetylcholins als Neurotransmitter hatte Dale bereits 1914
postuliert). Beide erhielten 1936 den Nobelpreis für Physiologie oder
Medizin "für ihre Entdeckungen bei der chemischen Übertragung der
Nervenimpulse" (Dale'sches Prinzip)
Der österreichische Pharmakologe Otto Loewi
konnte 1921 experimentell nachweisen, dass die parasympathische Wirkung
auf das Herz durch einen "Vagusstoff" vermittelt wird. Dieser wurde vom britischen Pharmakologen Sir Henry Dale als Acetylcholin identifiziert (eine
Rolle des Acetylcholins als Neurotransmitter hatte Dale bereits 1914
postuliert). Beide erhielten 1936 den Nobelpreis für Physiologie oder
Medizin "für ihre Entdeckungen bei der chemischen Übertragung der
Nervenimpulse" (Dale'sches Prinzip)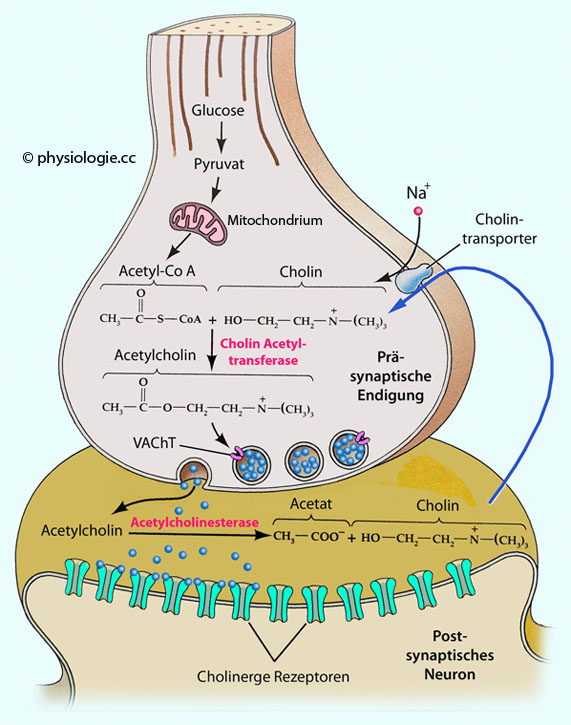 Abbildung: Stoffwechsel des Neurotransmitters Acetylcholin an cholinergen Synapsen
Abbildung: Stoffwechsel des Neurotransmitters Acetylcholin an cholinergen Synapsen Acetylcholin entsteht vorwiegend in cholinergen Nervenzellen (des zentralen und peripheren Nervensystems - an motorischen Endplatten sowie im autonomen Nervensystem) durch die Wirkung der Cholinacetyltransferase
(ChAT, Abbildung), die eine Acetylgruppe vom Coenzym Acetyl-CoA auf einen
Cholinrest überträgt. Die höchste Konzentration an Cholinacetyltransferase findet sich an
den präsynaptischen Endigungen der Axone, wo Acetylcholin
vesikulär gespeichert wird. Vesikuläre Acetylcholintransporter (VAChT) lagern pro cholinergem Vesikel 104 Acetylcholinmoleküle ein. Die neuronale Aufnahme von Cholin aus der extrazellulären Flüssigkeit ist der geschwindigkeitsbestimmende Schritt der Transmittersynthese.
Acetylcholin entsteht vorwiegend in cholinergen Nervenzellen (des zentralen und peripheren Nervensystems - an motorischen Endplatten sowie im autonomen Nervensystem) durch die Wirkung der Cholinacetyltransferase
(ChAT, Abbildung), die eine Acetylgruppe vom Coenzym Acetyl-CoA auf einen
Cholinrest überträgt. Die höchste Konzentration an Cholinacetyltransferase findet sich an
den präsynaptischen Endigungen der Axone, wo Acetylcholin
vesikulär gespeichert wird. Vesikuläre Acetylcholintransporter (VAChT) lagern pro cholinergem Vesikel 104 Acetylcholinmoleküle ein. Die neuronale Aufnahme von Cholin aus der extrazellulären Flüssigkeit ist der geschwindigkeitsbestimmende Schritt der Transmittersynthese. Freigesetztes Acetylcholin wirkt über ACh-Rezeptoren (sowohl prä- als auch postsynaptisch) und wird innerhalb von Millisekunden extrazellulär zu Acetat und Cholin abgebaut - durch Acetylcholinesterasen (AChE),
die an Basalmembranen zwischen prä- und postsynaptischen Zellen
befestigt und auf Nerven- und Muskelzellen sowie Erythrozyten vorhanden
sind. AChE sind hochaktiv (jedes Molekül kann pro Sekunde 5.103 Acetylcholinmoleküle spalten). Cholin wird von Neuronen wieder aufgenommen (Cholintransporter - dieser ist durch das Gift Hemicholinium
blockierbar). Nichtspezifische Enzyme (Pseudocholinesterasen,
Butyrylcholinesterasen) sind weiter verbreitet und bauen Acetylcholin
ebenfalls ab.
Freigesetztes Acetylcholin wirkt über ACh-Rezeptoren (sowohl prä- als auch postsynaptisch) und wird innerhalb von Millisekunden extrazellulär zu Acetat und Cholin abgebaut - durch Acetylcholinesterasen (AChE),
die an Basalmembranen zwischen prä- und postsynaptischen Zellen
befestigt und auf Nerven- und Muskelzellen sowie Erythrozyten vorhanden
sind. AChE sind hochaktiv (jedes Molekül kann pro Sekunde 5.103 Acetylcholinmoleküle spalten). Cholin wird von Neuronen wieder aufgenommen (Cholintransporter - dieser ist durch das Gift Hemicholinium
blockierbar). Nichtspezifische Enzyme (Pseudocholinesterasen,
Butyrylcholinesterasen) sind weiter verbreitet und bauen Acetylcholin
ebenfalls ab. 
 Es kann an postsynaptische Rezeptoren binden und entsprechende Effekte hervorrufenEs kann an präsynaptische Rezeptoren binden und weitere Freisetzung bremsen (negative Rückkopplung)Es kann durch Acetylcholinesterase abgebaut werden (Cholin kann von den Zellen wieder verwertet werden)
Es kann an postsynaptische Rezeptoren binden und entsprechende Effekte hervorrufenEs kann an präsynaptische Rezeptoren binden und weitere Freisetzung bremsen (negative Rückkopplung)Es kann durch Acetylcholinesterase abgebaut werden (Cholin kann von den Zellen wieder verwertet werden) Nikotonische Rezeptoren dienen insbesondere als Angriffspunkt
für Acetylcholin im Zentralnervensystem (z.B. Interneurone im Striatum, Nervenzellen im nucl. basalis Meynert) als auch für aus dem ZNS
austretende Fasern (Muskelzellen und autonom-nervöse Synapsen,
z.B. im Grenzstrang: präganglionär). Muskarinische
Nikotonische Rezeptoren dienen insbesondere als Angriffspunkt
für Acetylcholin im Zentralnervensystem (z.B. Interneurone im Striatum, Nervenzellen im nucl. basalis Meynert) als auch für aus dem ZNS
austretende Fasern (Muskelzellen und autonom-nervöse Synapsen,
z.B. im Grenzstrang: präganglionär). Muskarinische  Rezeptoren wirken in der Peripherie des parasympathischen Nervensystems (postganglionär). Rezeptoren (N-Acetylcholinrezeptoren, nAChR) sind nichtselektive Ionenkanäle, bestehend aus 5 Untereinheiten (mit zu ~40% identischen Aminosäuresequenzen), die um eine zentrale Pore (den Ionenkanal) angeordnet sind. Diese pentameren Kanäle stellen den Prototypen ligandengesteuerter Ionenkanäle dar.
Rezeptoren wirken in der Peripherie des parasympathischen Nervensystems (postganglionär). Rezeptoren (N-Acetylcholinrezeptoren, nAChR) sind nichtselektive Ionenkanäle, bestehend aus 5 Untereinheiten (mit zu ~40% identischen Aminosäuresequenzen), die um eine zentrale Pore (den Ionenkanal) angeordnet sind. Diese pentameren Kanäle stellen den Prototypen ligandengesteuerter Ionenkanäle dar. Abbildung).
Abbildung). Abbildung: Nikotinischer Acetylcholinrezeptor an motorischen Endplatten der Skelettmuskulatur (Endplattenpotentiale
- diese sind normalerweise so stark, dass sie auch dann noch
überschwellig auf die Muskelfaser wirken, wenn sie um 70-80% reduziert
werden) sowie in Ganglienzellen des autonomen Nervensystems (exzitatorische postsynaptische Potentiale EPSPs).
Die überschwellige Erregung einer postganglionären Nervenzelle
erfordert meist die gleichzeitige Anregung durch mehrere präganglionäre
Zellen (summativer Effekt, integrative Aktion; die meisten postganlionären Nervenzellen werden durch Axonfortsätze mehrerer präganglionärer Zellen versorgt).
Abbildung: Nikotinischer Acetylcholinrezeptor an motorischen Endplatten der Skelettmuskulatur (Endplattenpotentiale
- diese sind normalerweise so stark, dass sie auch dann noch
überschwellig auf die Muskelfaser wirken, wenn sie um 70-80% reduziert
werden) sowie in Ganglienzellen des autonomen Nervensystems (exzitatorische postsynaptische Potentiale EPSPs).
Die überschwellige Erregung einer postganglionären Nervenzelle
erfordert meist die gleichzeitige Anregung durch mehrere präganglionäre
Zellen (summativer Effekt, integrative Aktion; die meisten postganlionären Nervenzellen werden durch Axonfortsätze mehrerer präganglionärer Zellen versorgt).| Nikotinische Rezeptoren sind ligandengesteuerte Ionenkanäle |
Der Muskeltyp
besteht aus zwei α1-, einer ß1-, einer δ- und einer ε- (Erwachsenenform: in reifen,
innervierten Endplatten) bzw. γ- (fetale Form) Untereinheit. Über
diese Rezeptoren läuft die Übertragung an der motorischen Endplatte. Der Ganglientyp besteht aus α3- und ß4-Untereinheiten (Alpha-3 beta-4 nicotinic receptor). Man findet ihn in autonomen Ganglien, hauptsächlich postsynaptisch. Der ZNS-Typ besteht aus α4- und ß2- (Alpha-4 beta-2 nicotinic receptor)
(heteromerer ZNS-Typ), nur aus α7- (homomerer ZNS-Typ) oder α3- und
ß4-Untereinheiten (weiterer ZNS-Typ). Diese Rezeptoren finden sich prä-
und postsynaptisch vor allem im Gehirn. Pharmaka wirken auf diese Rezeptortypen unterschiedlich, wie die folgende Tabelle beispielhaft zeigt:
Pharmaka wirken auf diese Rezeptortypen unterschiedlich, wie die folgende Tabelle beispielhaft zeigt:| Einflüsse auf nikotinische Rezeptor-Subtypen (Beispiele) Nach Ritter / Flower / Henderson / Loke / MacEwan / Rang, Rang & Dale's Pharmacology, 9th ed. Elsevier 2020 |
|||
| Muskeltyp |
Ganglientyp |
ZNS-Typ |
|
| Agonisten |
Acetylcholin Carbachol Succinylcholin |
Acetylcholin Carbachol Nikotin |
Nikotin Epibatidin Acetylcholin |
| Antagonisten |
Tubocurarin Pancuronium Bungarotoxin |
Mecamylamin Trimetaphan Hexamethonium |
Mecamylamin Methylaconitin |
 Abbildung: Wichtigste pharmakologische Wirkungen auf cholinerge Übertragung
Abbildung: Wichtigste pharmakologische Wirkungen auf cholinerge Übertragung| Muskarinische Rezeptoren sind G-Protein-gekoppelt |
Abbildung oben): Rasche Depolarisierung (initiale EPSPs, die zu
Aktionspotentialen führen können); Hyperpolarisierung (IPSPs, durch
M2-Rezeptoren vermittelt); sekundäre, langsame EPSPs, durch M1-Rezeptoren
vermittelt; und späte langsame EPSPs, mediiert durch verschiedene
Peptide / Kotransmitter (wie GnRH, Substanz P, Angiotensin, CGRP, VIP, NPY, Enkephaline).in ZNS (Großhirnrinde, Hippocampus, Striatum),in peripheren Neuronen (autonome Ganglien) - hier fördern sie über Senkung der K+-Leitfähigkeit und langsame Depolarisierung (exzitatorische postsynaptische Potentiale) die Erregbarkeit,in Belegzellen der Magenschleimhaut (Salzsäureproduktion durch Vagusaktivität), in exokrinen Drüsen (Speichel-, Tränen- u.a.). in den Herzvorhöfen - über erhöhte K+-Leitfähigkeit (Hyperpolarisierung) und Inhibition von Ca++-Kanälen - Wirkungen des Parasympathikus auf Herzqualitäten: Sie verlangsamen die Geschwindigkeit von Depolarisation (negativ chronotrop: Sinusknoten!)
und Erregungsleitung (negativ dromotrop: AV-Knoten!) - starke Reizung kann zu Asystolie (Herzstillstand) führen (z.B. beim "Bolustod"). In den Vorhöfen
senken sie auch die Kontraktionskraft (negativ inotrop), in den Herzkammern nur indirekt (über Wirkung auf sympathische Nervenfasern). Darüber hinaus bewirkt Reizung von M2-Rezeptoren Kontraktion der Harnblase (Detrusion) und regen die Ureterperistaltik an. Im Gehirn sind sie weit verbreitet, u.a. im basalen Vorderhirn, Thalamus. Sie wirken hier als Autorezeptoren,
d.h. sie hemmen weitere Freisetzung des Neurotransmitters aus der
Zelle, die diesen sezerniert (negative Rückkopplung) - vor allem in der
Hirnrinde und im Hippocampus.In der Zellmembran postganglionärer parasympathischer Neurone hemmen sie weitere ACh-Freisetzung (Autoinhibition). regen sie die Sekretion in Speichel-, Bronchial- (Schleimbildung) oder Schweißdrüsen an,bewirken Kontraktion glatter Muskelzellen ("glattmuskulär", z.B. Bronchokonstriktion, angeregte Peristaltik und Tonuserhöhung im Darm, Ureterperistaltik und gesteigerter Blasendruck),aber Dilatation von Blutgefäßen: Stimulierung von M3-Rezeptoren
auf Endothelzellen veranlasst diese zur Bildung von NO, dieses
diffundiert an benachbarte Muskelzellen und bewirkt Vasodilatation.M3-Rezeptoren vermitteln die meisten
postganglionär-parasympathischen Effekte, z.B. im Auge (kann den Augeninnendruck reduzieren), Sekretion im gastrointestinalen
System (Salivation, Magensaftbildung), Schweiß- und TränenbildungM3-Rezeptoren kommen auch im Gehirn vor (Großhirnrinde, Hippocampus, Thalamus, Hinstamm) und beteiligen sich u.a. an der Auslösung des
Brechreflexes. M4: ZNS (Großhirnrinde, Hippocampus, Striatum) - Förderung der Lokomotion. M4-Rezeptoren wirken als Autorezeptoren (negative Rückkopplung). So hemmen sie die Acetylcholinfreisetzung im Striatum. M5: Substantia nigra, Auge (Iris, Ziliarkörper: Miosis, Akkommodation), Speicheldrüsen (Sekretion). Alle muskarinischen Rezeptoren werden durch Acetylcholin angeregt und durch Atropin blockiert. Pirenzepin blockiert selektiv M1-Rezeptoren, Gallamin M2-Rezeptoren. Abbildung: Muskarinischer Acetylcholinrezeptor
Abbildung: Muskarinischer Acetylcholinrezeptor GIRK, G protein-coupled inwardly rectifying K channels MAPK, mitogenaktivierte Proteinkinasen PLC, Phosphoinositid-Phospholipase C, verwandelt PIP2 zu IP3 und DAG VDCC, Voltage dependent calcium channel Über second-messenger-Mechanismen s. auch dort Komplette Auflistung der Wirkungen im muskarinergen System s. dort
GIRK, G protein-coupled inwardly rectifying K channels MAPK, mitogenaktivierte Proteinkinasen PLC, Phosphoinositid-Phospholipase C, verwandelt PIP2 zu IP3 und DAG VDCC, Voltage dependent calcium channel Über second-messenger-Mechanismen s. auch dort Komplette Auflistung der Wirkungen im muskarinergen System s. dort
Signalwege cholinerger Übertragung Nach Boron / Boulpaep: Concise Medical Physiology, Elsevier 2021 |
|||||
| Rezeptor |
Agonisten |
Antagonisten |
G-Protein |
Enzym |
second messenger |
| N1- nikotinisch |
Acetylcholin Nicotin |
Tubocuranin Bungarotoxin |
- |
- |
- |
| N2- nikotinisch |
Acetylcholin Nicotin |
Hexamethonium |
- |
- |
- |
| M1/3/5- muskarinisch |
Acetylcholin Muscarin |
Atropin |
Gαq |
PLC |
IP3 / DAG |
| M2/4- muskarinisch |
Acetylcholin Muscarin |
Atropin | Gαq, Gαo |
Adenylylcyclase |
↓[cAMP] |
Abbildung.  Abbildung: Acetylcholinsynthese und -freisetzung in einer Varikosität Präsynaptische Blockade des Acetylcholinmechanismus: Der Natrium-Cholin- Cotransporter und damit die Cholinaufnahme kann durch Hemicholinium, der vesikuläre Acetylcholintransporter und damit die Aufnahme in Vesikel durch Vesamicol, die Exozytose und damit Freisetzung von Acetylcholin durch Botulinumtoxin gehemmt werden.Die Entdeckung des Muscarins (1869) geht auf den deutschen Pharmakologen Oswald Schmiedeberg zurück: Es wurde als ein Wirkstoff des Fliegenpilzes (Amanita muscarina) identifiziert. Henry Dale nannte Wirkungen des Acetylcholins "muscarine actions" (1914)
Abbildung: Acetylcholinsynthese und -freisetzung in einer Varikosität Präsynaptische Blockade des Acetylcholinmechanismus: Der Natrium-Cholin- Cotransporter und damit die Cholinaufnahme kann durch Hemicholinium, der vesikuläre Acetylcholintransporter und damit die Aufnahme in Vesikel durch Vesamicol, die Exozytose und damit Freisetzung von Acetylcholin durch Botulinumtoxin gehemmt werden.Die Entdeckung des Muscarins (1869) geht auf den deutschen Pharmakologen Oswald Schmiedeberg zurück: Es wurde als ein Wirkstoff des Fliegenpilzes (Amanita muscarina) identifiziert. Henry Dale nannte Wirkungen des Acetylcholins "muscarine actions" (1914) sind Bausteine von Nukleinsäuren (Nukleoside wie Adenosin, Nukleotide
wie ATP und ADP). ATP findet sich in allen synaptischen Vesikeln und
wird von der präsynaptischen Nervenendigung zusammen mit "klassischen"
Neurotransmittern freigesetzt, und Neuronen reagieren auf extrazellulär
auftretende Purine mit elektrischen Reaktionen: So löst ATP an
motorischen, sensorischen und autonomen Nervenzellen exzitatorische
Effekte (Depolarisationen) aus.
sind Bausteine von Nukleinsäuren (Nukleoside wie Adenosin, Nukleotide
wie ATP und ADP). ATP findet sich in allen synaptischen Vesikeln und
wird von der präsynaptischen Nervenendigung zusammen mit "klassischen"
Neurotransmittern freigesetzt, und Neuronen reagieren auf extrazellulär
auftretende Purine mit elektrischen Reaktionen: So löst ATP an
motorischen, sensorischen und autonomen Nervenzellen exzitatorische
Effekte (Depolarisationen) aus. 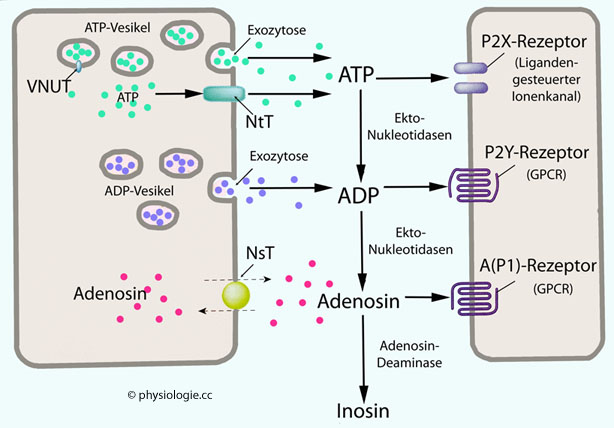 Abbildung: Purine und ihre RezeptorenAbbildung): Adenosinrezeptoren (A1, A2A, A2B, A3- früher als P1-Rezeptoren bezeichnet) sind GPCR, die über cAMP oder direkte Wirkung auf Ca++- und K+-Kanäle wirken. Metabotrope P2Y-Rezeptoren (P2Y1-14) sind metabotrop (GPCR) und wirken über PLC oder cAMP. Sie sprechen vor allem auf ATP an, auch auf ADP, AMP, UTP. Ionotrope P2X-Rezeptoren (P2X1-7) sind
trimere ATP-gesteuerte Ionenkanäle (mit je 2 membrandurchspannenden α-Helices), deren Aktivierung Na+ und Ca++
durch die Membran passieren lässt. Depolarisierung aktiviert
calciumabhängige Mechanismen in der Zelle. P2X-Rezeptoren wirken u.a.
als
Gefahrendetektoren (chemisch, Hitze) im peripheren sensorischen
Nervensystem.
Abbildung: Purine und ihre RezeptorenAbbildung): Adenosinrezeptoren (A1, A2A, A2B, A3- früher als P1-Rezeptoren bezeichnet) sind GPCR, die über cAMP oder direkte Wirkung auf Ca++- und K+-Kanäle wirken. Metabotrope P2Y-Rezeptoren (P2Y1-14) sind metabotrop (GPCR) und wirken über PLC oder cAMP. Sie sprechen vor allem auf ATP an, auch auf ADP, AMP, UTP. Ionotrope P2X-Rezeptoren (P2X1-7) sind
trimere ATP-gesteuerte Ionenkanäle (mit je 2 membrandurchspannenden α-Helices), deren Aktivierung Na+ und Ca++
durch die Membran passieren lässt. Depolarisierung aktiviert
calciumabhängige Mechanismen in der Zelle. P2X-Rezeptoren wirken u.a.
als
Gefahrendetektoren (chemisch, Hitze) im peripheren sensorischen
Nervensystem. Die Bestimmung der
Purinkonzentration in Körperflüssigkeiten (mittels HPLC) hat in speziellen Fällen klinisch-diagnostische Bedeutung (Stoffwechselstörungen).
Die Bestimmung der
Purinkonzentration in Körperflüssigkeiten (mittels HPLC) hat in speziellen Fällen klinisch-diagnostische Bedeutung (Stoffwechselstörungen).  Abbildung: Kotransmission an autonom-nervöser Varikosität
(1) Purinerg: Der am schnellsten eintretende Kontraktionseffekt
ist bedingt durch den Einstrom von Calciumionen, deren Kanäle durch
Depolarisierung geöffnet wurden, die wiederum via Kationeneinstrom
durch purinerg angeregte Ionenkanäle erfolgt. (2) Adrenerg: Die Anregung von α1-Rezeptoren aktiviert (über Gq-Protein) IP3, das zu endoplasmatischen Vesikeln diffundiert und hier Calciumkanäle öffnet.
(3) Peptiderg: Über Y-Rezeptorern bewirkt Neuropeptid Y eine
langsamer auftretende, aber länger anhaltende Steigerung des
zytoplasmatischen Calciumspiegels in den Zielzellen. Abbildung oben): Neuronen nützen gleichzeitig mehrere Klassen von
Molekülen für die interzelluläre Kommunikation. ATP wird von postganglionär-sympathischen Fasern
zusammen mit Noradrenalin aus synaptischen Vesikeln ausgeschüttet.
Direkt am glatten Gefäßmuskel wirkt es vasokonstriktorisch.
Abbildung: Kotransmission an autonom-nervöser Varikosität
(1) Purinerg: Der am schnellsten eintretende Kontraktionseffekt
ist bedingt durch den Einstrom von Calciumionen, deren Kanäle durch
Depolarisierung geöffnet wurden, die wiederum via Kationeneinstrom
durch purinerg angeregte Ionenkanäle erfolgt. (2) Adrenerg: Die Anregung von α1-Rezeptoren aktiviert (über Gq-Protein) IP3, das zu endoplasmatischen Vesikeln diffundiert und hier Calciumkanäle öffnet.
(3) Peptiderg: Über Y-Rezeptorern bewirkt Neuropeptid Y eine
langsamer auftretende, aber länger anhaltende Steigerung des
zytoplasmatischen Calciumspiegels in den Zielzellen. Abbildung oben): Neuronen nützen gleichzeitig mehrere Klassen von
Molekülen für die interzelluläre Kommunikation. ATP wird von postganglionär-sympathischen Fasern
zusammen mit Noradrenalin aus synaptischen Vesikeln ausgeschüttet.
Direkt am glatten Gefäßmuskel wirkt es vasokonstriktorisch.
 < ATP
wird durch oxidative Phosphorylierung aus ADP gebildet. Es regt die
glatte Muskulatur in Arterienwänden, Blase und ductus deferens an. Abbildung oben). ATP ist ein Kotransmitter im sympathischen System (mit
Noradrenalin), in manchen cholinergen Nerven, und im Darmnervensystem. ATP wirkt auch schmerz- und
entzündungsauslösend.
< ATP
wird durch oxidative Phosphorylierung aus ADP gebildet. Es regt die
glatte Muskulatur in Arterienwänden, Blase und ductus deferens an. Abbildung oben). ATP ist ein Kotransmitter im sympathischen System (mit
Noradrenalin), in manchen cholinergen Nerven, und im Darmnervensystem. ATP wirkt auch schmerz- und
entzündungsauslösend. 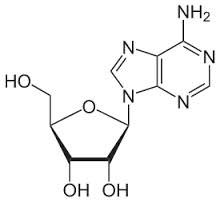 < Extrazelluläres Adenosin
stammt einerseits aus dem (extrazellulären) Abbau von Transmitter-ATP,
andererseits aus Zellen, die es bei erhöhtem (intrazellulärem)
ATP-Abbau an ihre Umgebung abgeben - z.B. aktive Muskelzellen, Zellen der macula densa (tubulo-glomeruläre Rückkopplung), Zellen im Gehirn.
< Extrazelluläres Adenosin
stammt einerseits aus dem (extrazellulären) Abbau von Transmitter-ATP,
andererseits aus Zellen, die es bei erhöhtem (intrazellulärem)
ATP-Abbau an ihre Umgebung abgeben - z.B. aktive Muskelzellen, Zellen der macula densa (tubulo-glomeruläre Rückkopplung), Zellen im Gehirn. Abbildung: Adenosinrezeptor Abbildung) u.a. als "homöostatischer
Modulierer" in Herz (A1: bremst Erregungsleitung, anti-dysrhythmischer
Effekt; A1 und A2A:
koronare Vasodilatation; wahrscheinlich sind alle Adenosinrezeptortypen
an den kardialen Effekten des Adenosins beteiligt) und Lunge (bronchokonstriktorische Wirkung? Adenosin wirkt auch hier über mehrere Rezeptortypen, mit unterschiedlicher Wirkung). An Gefäßmuskelzellen bewirkt es Dilatation und Durchblutungssteigerung über A2A-Rezeptoren → cAMP → Proteinkinase A (→ Erniedrigung zytosolisches [Ca++], verstärkter K-Ausstrom), über A1-Rezeptoren → Öffnung KATP-Kanäle, und über Reduktion cAMP-stimulierter Noradrenalinfreisetzung sympathischer Varikositäten. Im Gehirn wird Adenosin bei erhöhtem Energieverbrauch von den Nervenzellen freigesetzt. Es wirkt allgemein inhibierend (Sedierung, Krampflösung,
Schmerzstillung) und hat einen protektiven Effekt auf das Nervengewebe
(Sicherheitsmechanismus, z.B. bei Sauerstoffmangel oder Übererregung).
Die Stimulierende Wirkung von Xanthinderivaten wie Coffein beruht auf einer Hemmung von P1- bzw. A2A-Rezeptoren, was ihren Weckeffekt zumindest teilweise erklärt.
Abbildung: Adenosinrezeptor Abbildung) u.a. als "homöostatischer
Modulierer" in Herz (A1: bremst Erregungsleitung, anti-dysrhythmischer
Effekt; A1 und A2A:
koronare Vasodilatation; wahrscheinlich sind alle Adenosinrezeptortypen
an den kardialen Effekten des Adenosins beteiligt) und Lunge (bronchokonstriktorische Wirkung? Adenosin wirkt auch hier über mehrere Rezeptortypen, mit unterschiedlicher Wirkung). An Gefäßmuskelzellen bewirkt es Dilatation und Durchblutungssteigerung über A2A-Rezeptoren → cAMP → Proteinkinase A (→ Erniedrigung zytosolisches [Ca++], verstärkter K-Ausstrom), über A1-Rezeptoren → Öffnung KATP-Kanäle, und über Reduktion cAMP-stimulierter Noradrenalinfreisetzung sympathischer Varikositäten. Im Gehirn wird Adenosin bei erhöhtem Energieverbrauch von den Nervenzellen freigesetzt. Es wirkt allgemein inhibierend (Sedierung, Krampflösung,
Schmerzstillung) und hat einen protektiven Effekt auf das Nervengewebe
(Sicherheitsmechanismus, z.B. bei Sauerstoffmangel oder Übererregung).
Die Stimulierende Wirkung von Xanthinderivaten wie Coffein beruht auf einer Hemmung von P1- bzw. A2A-Rezeptoren, was ihren Weckeffekt zumindest teilweise erklärt.  Methylxanthine Weiters wirkt Adenosin auf Lipogenese / Lipolyse (Fettgewebe), Glukoneogenese / Glykogenolyse (Leber) sowie Glucoseaufnahme (Muskulatur) - unterschiedlich je nach involvierten Rezeptoren (A1, A2A, A2B). Amine
sind Derivate aromatischer Aminosäuren (wie Tyrosin, Trypotophan,
Histidin), die u.a. als Neurotransmitter wirken (z.B. Katecholamine,
Serotonin, Histamin). Zu biogenen Aminen gehören Katecholamine (Adrenalin, Noradrenalin, Dopamin), Histamin, Serotonin, Melatonin.
Methylxanthine Weiters wirkt Adenosin auf Lipogenese / Lipolyse (Fettgewebe), Glukoneogenese / Glykogenolyse (Leber) sowie Glucoseaufnahme (Muskulatur) - unterschiedlich je nach involvierten Rezeptoren (A1, A2A, A2B). Amine
sind Derivate aromatischer Aminosäuren (wie Tyrosin, Trypotophan,
Histidin), die u.a. als Neurotransmitter wirken (z.B. Katecholamine,
Serotonin, Histamin). Zu biogenen Aminen gehören Katecholamine (Adrenalin, Noradrenalin, Dopamin), Histamin, Serotonin, Melatonin.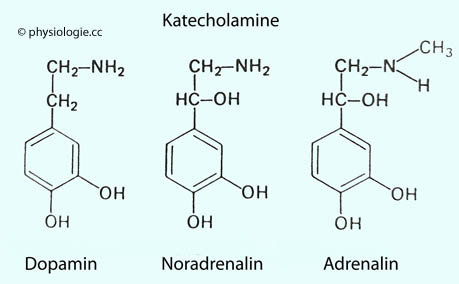 Speicherung und Freisetzung Abbau Referenzwerte Dopamin Adrenalin, Noradrenalin α- und ß-Rezeptoren Katecholamine
sind biogene Amine, die durch mehrfache enzymatische Schritte - über
DOPA - aus der Aminosäure Tyrosin gebildet werden ( Abbildung unten). Sie bestehen aus einer Katecholgruppe (doppelt hydroxylierter Phenolring)
und einer Amino-Seitenkette. Wichtigste Vertreter sind Dopamin,
Noradrenalin und Adrenalin. Sie werden von adrenergen Nervenfasern freigesetzt, insbesondere deren Varikositäten.
Speicherung und Freisetzung Abbau Referenzwerte Dopamin Adrenalin, Noradrenalin α- und ß-Rezeptoren Katecholamine
sind biogene Amine, die durch mehrfache enzymatische Schritte - über
DOPA - aus der Aminosäure Tyrosin gebildet werden ( Abbildung unten). Sie bestehen aus einer Katecholgruppe (doppelt hydroxylierter Phenolring)
und einer Amino-Seitenkette. Wichtigste Vertreter sind Dopamin,
Noradrenalin und Adrenalin. Sie werden von adrenergen Nervenfasern freigesetzt, insbesondere deren Varikositäten.| Charakteristika von Katecholaminen |
| Synthese durch enzymatische Aktivität aus Tyrosin |
| Speicherung in Vesikeln |
| Regulation über Sekretion (Exozytose) und Enzyme (gesteuerte Zwischenschritte) |
| Transport im Blut frei und in locker eiweißgebundener Form |
| Wirkung über adrenerge (membranständige) Rezeptoren |
| Gabe als Aerosol möglich (broncholytische Wirkung), Analoga auch oral |
Abbildung).
 Abbildung: Katecholamine entstehen aus Tyrosin Das pflanzliche Alkaloid Reserpin hemmt diesen Transport durch VMAT und damit die Aufnahme von Monoaminen (Dopamin, Noradrenalin, Adrenalin, Serotonin) in Vesikel: Noradrenalin verbleibt im Zytoplasma und wird durch MAO abgebaut (s. unten). Das führt zu einer Transmitter-Entspeicherung sympathischer Fasern und monoaminerger Neurone im Gehirn, die sympathische Wirkung schwächt sich ab. Abbildung zeigt Details:
Abbildung: Katecholamine entstehen aus Tyrosin Das pflanzliche Alkaloid Reserpin hemmt diesen Transport durch VMAT und damit die Aufnahme von Monoaminen (Dopamin, Noradrenalin, Adrenalin, Serotonin) in Vesikel: Noradrenalin verbleibt im Zytoplasma und wird durch MAO abgebaut (s. unten). Das führt zu einer Transmitter-Entspeicherung sympathischer Fasern und monoaminerger Neurone im Gehirn, die sympathische Wirkung schwächt sich ab. Abbildung zeigt Details: Abbildung: Adrenerge Synapse Über den Synaptobrevin- Syntaxin- Mechanismus s. auch dort Acetylcholin (muskarinerg), Noradrenalin (über α2- autoinhibitorisch - und ß2-Rezeptoren - verstärkend auf die NA-Freisetzung), Dopamin, Serotonin, Angiotensin II, Prostaglandine, Purine, Neuropeptide u.a.
Abbildung: Adrenerge Synapse Über den Synaptobrevin- Syntaxin- Mechanismus s. auch dort Acetylcholin (muskarinerg), Noradrenalin (über α2- autoinhibitorisch - und ß2-Rezeptoren - verstärkend auf die NA-Freisetzung), Dopamin, Serotonin, Angiotensin II, Prostaglandine, Purine, Neuropeptide u.a. 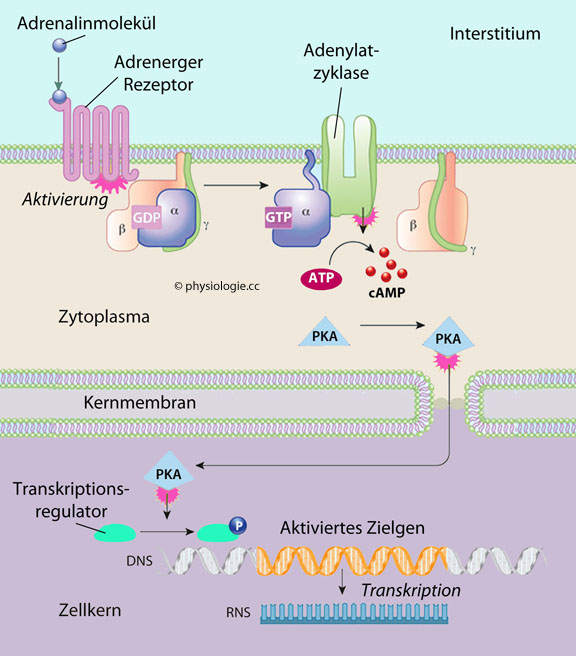 Abbildung: Adrenalin: Signaltransduktionskaskade mit cAMP Der Abbau der Katecholamine erfolgt mittels zweier Enzyme (Abbildung): Monoaminooxidase (MAO)
kommt in der äußeren Mitochondrienmembran der meisten
Zellen vor. MAO-A findet sich vor allem in Neuronen und Gliazellen
(Astrozyten), weiters in Leberzellen, pulmonalen Endothelzellen, Zellen
des Magen-Darm-Trakts und der Plazenta; MAO-B in Thrombozyten, auch in
Nerven- und Gliazellen. Die
entstehenden Aldehyde werden in der Peripherie durch ADH dehydrogeniert
(und entgiftet). Die Catechol-O-Methlytransferase (COMT)
ist ebenfalls weit verbreitet - wie in Zellen des Nebennierenmarks
(nicht in
katecholaminergen Neuronen). Es gibt eine zytoplasmatische lösliche
(S-COMT, soluble) und eine membrangebundene Form (MB-COMT, membrane-bound). COMT fügt eine Methylgruppe an das
Katecholaminmolekül an. So macht sie aus
Adrenalin Metanephrin und aus Noradrenalin Normetanephrin; 3,4-Dihydroxymandelsäure methyliert sie zu Vanillinmandelsäure.
Abbildung: Adrenalin: Signaltransduktionskaskade mit cAMP Der Abbau der Katecholamine erfolgt mittels zweier Enzyme (Abbildung): Monoaminooxidase (MAO)
kommt in der äußeren Mitochondrienmembran der meisten
Zellen vor. MAO-A findet sich vor allem in Neuronen und Gliazellen
(Astrozyten), weiters in Leberzellen, pulmonalen Endothelzellen, Zellen
des Magen-Darm-Trakts und der Plazenta; MAO-B in Thrombozyten, auch in
Nerven- und Gliazellen. Die
entstehenden Aldehyde werden in der Peripherie durch ADH dehydrogeniert
(und entgiftet). Die Catechol-O-Methlytransferase (COMT)
ist ebenfalls weit verbreitet - wie in Zellen des Nebennierenmarks
(nicht in
katecholaminergen Neuronen). Es gibt eine zytoplasmatische lösliche
(S-COMT, soluble) und eine membrangebundene Form (MB-COMT, membrane-bound). COMT fügt eine Methylgruppe an das
Katecholaminmolekül an. So macht sie aus
Adrenalin Metanephrin und aus Noradrenalin Normetanephrin; 3,4-Dihydroxymandelsäure methyliert sie zu Vanillinmandelsäure.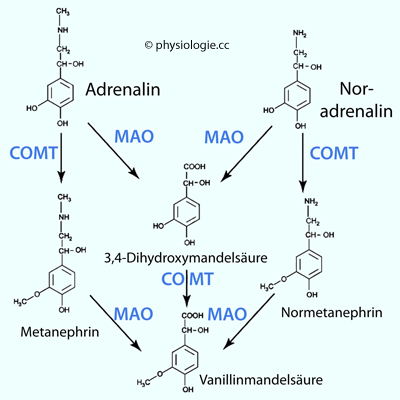 Abbildung: Abbau von Adrenalin und Noradrenalin
Abbildung: Abbau von Adrenalin und Noradrenalin Katecholamine Adrenalin s. dort Noradrenalin s. dort
Dopamin s. dort Ausscheidung Homovanillinsäure, Vanillinmandelsäure s. dort
Katecholamine Adrenalin s. dort Noradrenalin s. dort
Dopamin s. dort Ausscheidung Homovanillinsäure, Vanillinmandelsäure s. dort| Katecholamine haben eine sehr kurze Halbwertszeit (Sekunden bis Minuten) |
Rezeptoren Wirkungen Zur Synthese des Dopamins vgl. oben. Dopamin ist der Vorläufer von Adrenalin und Noradrenalin. Dopamin entsteht (präsynaptisch) aus Tyrosin - das eisenhaltige Enzym Phenylalaninhydroxylase stellt es (vor allem in der Leber) aus der essentiellen Aminosäure Phenylalanin her - durch die konsekutive Wirkung zweier Enzyme (Tyrosinhydroxylase → DOPA, aromatische Aminosäure-Decarboxylase). Die Aktivität der Tyrosinhydroxylase ist (wie auch bei der Synthese anderer Katecholamine) der limitierende Schritt. Anschließend wird Dopamin mittels des vesikulären Monoaminotransporters VMAT2 in Vesikel gespeichert ( Abbildung).
Dopamin wird (wie andere Monoamine) nach seiner Freisetzung in den
Extrazellulärraum zum Teil von den präsynaptischen Zellen wieder aufgenommen (DAT: dopamine transporter) und wiederverwertet. Der Abbau erfolgt über MAO und COMT (s. oben) teils zu 3,4-Dihydroxyphenylessigsäure (DOPAC), teils bis zur Homovanillinsäure. Diese Abbauprodukte werden aus der Zelle exportiert und mit dem Harn ausgeschieden. Abbildung), die nach ihrem Wirkungsmechanismus eingeteilt werden in die D1-Subfamilie (regen die Adenylylcyclase an) und die D2-Subfamilie (hemmen die Adenylylcyclase): Abbildung: Dopaminrezeptor Zur D1-Subfamilie (D1/5-Gruppe) gehören D1- (der am stärksten exprimierte Dopaminrezeptor, im ZNS im Neostriatum am intensivsten vertreten) und D5-Rezeptoren (GS-Protein → Adenylylcyclase → cAMP → Aktivierung intrazellulärer Proteine ... Nervenzelle wird
aktiviert). Hierher gehören u.a. Rezeptoren auf renalen Blutgefäßen,
die Vasodilatation vermitteln. Zur D2-Subfamilie (D2/3/4-Gruppe) gehören D2- (überall im Gehirn exprimiert), D3- (nur im limbischen System) und die besonders polymorphen D4-Rezeptoren (GI-Protein → Hemmung der Adenylylcyclase → weniger cAMP; Aktivierung von
Kalium-Kanälen ... Ruhepotential wird stabilisiert, Nervenzelle gehemmt). Rezeptoren dieser Gruppe hemmen cholinerge Interneurone im Striatum sowie die Prolactinfreisetzung im Hypophysenvorderlappen.
Abbildung: Dopaminrezeptor Zur D1-Subfamilie (D1/5-Gruppe) gehören D1- (der am stärksten exprimierte Dopaminrezeptor, im ZNS im Neostriatum am intensivsten vertreten) und D5-Rezeptoren (GS-Protein → Adenylylcyclase → cAMP → Aktivierung intrazellulärer Proteine ... Nervenzelle wird
aktiviert). Hierher gehören u.a. Rezeptoren auf renalen Blutgefäßen,
die Vasodilatation vermitteln. Zur D2-Subfamilie (D2/3/4-Gruppe) gehören D2- (überall im Gehirn exprimiert), D3- (nur im limbischen System) und die besonders polymorphen D4-Rezeptoren (GI-Protein → Hemmung der Adenylylcyclase → weniger cAMP; Aktivierung von
Kalium-Kanälen ... Ruhepotential wird stabilisiert, Nervenzelle gehemmt). Rezeptoren dieser Gruppe hemmen cholinerge Interneurone im Striatum sowie die Prolactinfreisetzung im Hypophysenvorderlappen.| Dopaminerge Rezeptoren sind metabotrop und G-Protein-gekoppelt |
Signalwege dopaminerger Übertragung Modifiziert nach Ritter / Flower / Henderson / Loke / MacEwan / Rang, Rang & Dale's Pharmacology, 9th ed. Elsevier 2020 |
|||
| Rezeptor |
G-Prot. |
second messenger | Wirkungen |
| D1-Typ (D1, D5) |
Gs | ↑[cAMP] | Postsynaptische Inhibition |
| D2-Typ (D2, D3, D4) |
Gi | ↓[cAMP] | K+-Kanäle + Ca++-Kanäle - Prä- und postsynaptische Inhibition |
 Abbildung: Dopaminerge Schaltstelle ALDH = Aldehyddehydrogenase COMT = Catechol-O-Methlytransferase
DA = Dopamin DOPA = Dihydroxyphenylalanin DOPAC =
3,4-Dihydroxyphenylessigsäure MAO = Monoaminooxydase TH =
Tyrosinhydroxylase Über dopaminerge Systeme im Gehirn s. auch dort. In
der Niere wirkt Dopamin als auto- und parakriner Neurotransmitter. Es
bindet an Rezeptoren beider Subfamilien (s. unten) und steigert die
Natriurese (Hemmung verschiedener Na+-Transporter, wie die basolaterale Na/K-Pumpe und der luminale Na/H-Austauscher). Über D1-Rezeptoren steigert, über D3-Rezeptoren senkt es die Reninbildung. Im Nebennierenmark wird es als Vorstufe zur Adrenalinbildung gespeichert. Dopamin kommt im Darmnervensystem vor (dopaminerge Neurone, Dopamin als Vorstufe für andere Katecholamine). Dopamin
moduliert den Kontraktionszustand peripherer Gefäße, die
Nierendurchblutung und die Herzfunktion (niedrige Dosen reduzieren über
D1-Rezeptoren Gefäßtonus und Nachlast, mittlere Dosen
steigern über ß-Rezeptoren die Schlagkraft, hohe Dosen können über
α-Rezeptoren vasokonstriktorisch und damit blutdruckerhöhend wirken). Abbildung oben: Die Wiederaufnahme von Noradrenalin in die präsynaptische Nervenzelle durch NET kann durch Kokain sowie durch trizyklische Antidepressiva gehemmt werden. Das verlängert die Katecholaminwirkung am synaptischen Spalt.
Abbildung: Dopaminerge Schaltstelle ALDH = Aldehyddehydrogenase COMT = Catechol-O-Methlytransferase
DA = Dopamin DOPA = Dihydroxyphenylalanin DOPAC =
3,4-Dihydroxyphenylessigsäure MAO = Monoaminooxydase TH =
Tyrosinhydroxylase Über dopaminerge Systeme im Gehirn s. auch dort. In
der Niere wirkt Dopamin als auto- und parakriner Neurotransmitter. Es
bindet an Rezeptoren beider Subfamilien (s. unten) und steigert die
Natriurese (Hemmung verschiedener Na+-Transporter, wie die basolaterale Na/K-Pumpe und der luminale Na/H-Austauscher). Über D1-Rezeptoren steigert, über D3-Rezeptoren senkt es die Reninbildung. Im Nebennierenmark wird es als Vorstufe zur Adrenalinbildung gespeichert. Dopamin kommt im Darmnervensystem vor (dopaminerge Neurone, Dopamin als Vorstufe für andere Katecholamine). Dopamin
moduliert den Kontraktionszustand peripherer Gefäße, die
Nierendurchblutung und die Herzfunktion (niedrige Dosen reduzieren über
D1-Rezeptoren Gefäßtonus und Nachlast, mittlere Dosen
steigern über ß-Rezeptoren die Schlagkraft, hohe Dosen können über
α-Rezeptoren vasokonstriktorisch und damit blutdruckerhöhend wirken). Abbildung oben: Die Wiederaufnahme von Noradrenalin in die präsynaptische Nervenzelle durch NET kann durch Kokain sowie durch trizyklische Antidepressiva gehemmt werden. Das verlängert die Katecholaminwirkung am synaptischen Spalt.| Alle Adrenozeptoren sind metabotrop, sie wirken über G-Proteine |
 Sympathische (postsynaptische) Nervenfasern geben vorwiegend Noradrenalin ab.
Sympathische (postsynaptische) Nervenfasern geben vorwiegend Noradrenalin ab. Bei sympathischen Fasern beträgt das Verhältnis Noradrenalin / Adrenalin etwa 20 zu 1 (Noradrenalin ~95%).
Das Nebennierenmark verfügt über Phenylethanolamin-N-Methyltransferase,
somit kann es Noradrenalin ("Nor" = N ohne Radikal,
d.h. ohne Methylgruppe) zu Adrenalin umbauen. In der Nebenniere beträgt das Verhältnis Noradrenalin / Adrenalin etwa 1 zu 4 (Adrenalin ~80%). Extrazelluläres
Noradrenalin, das nicht präsynaptisch wiederaufgenommen wurde (über NET: Norepinephrine transporter), gelangt
in den Kreislauf , wird von extraneuronalen Zellen über Transporter
(ENTs: Extraneuronal transporters; sowie organische Kationentransporter) aufgenommen und abgebaut - zu 3-Methoxy-4-hydroxy-phenylglykol (MHPG)
- und u.a. in dieser Form im Harn ausgeschieden. Die Leber ist das
Hauptorgan des extraneuronalen Abbaus, das benutzte Enzym ist die
Katechol-O-Methlytransferase (COMT).
Bei sympathischen Fasern beträgt das Verhältnis Noradrenalin / Adrenalin etwa 20 zu 1 (Noradrenalin ~95%).
Das Nebennierenmark verfügt über Phenylethanolamin-N-Methyltransferase,
somit kann es Noradrenalin ("Nor" = N ohne Radikal,
d.h. ohne Methylgruppe) zu Adrenalin umbauen. In der Nebenniere beträgt das Verhältnis Noradrenalin / Adrenalin etwa 1 zu 4 (Adrenalin ~80%). Extrazelluläres
Noradrenalin, das nicht präsynaptisch wiederaufgenommen wurde (über NET: Norepinephrine transporter), gelangt
in den Kreislauf , wird von extraneuronalen Zellen über Transporter
(ENTs: Extraneuronal transporters; sowie organische Kationentransporter) aufgenommen und abgebaut - zu 3-Methoxy-4-hydroxy-phenylglykol (MHPG)
- und u.a. in dieser Form im Harn ausgeschieden. Die Leber ist das
Hauptorgan des extraneuronalen Abbaus, das benutzte Enzym ist die
Katechol-O-Methlytransferase (COMT). | α1-Adrenozeptoren aktivieren Phospholipase C |
 Abbildung: Katecholaminrezeptoren
Abbildung: KatecholaminrezeptorenSympathische Neurotransmission Modifiziert nach Ritter / Flower / Henderson / Loke / MacEwan / Rang, Rang & Dale's Pharmacology, 9th ed. Elsevier 2020 |
|||||
| α1-Rezeptoren | α2-Rezeptoren | ß1-Rezeptoren | ß2-Rezeptoren | ß3-Rezeptoren | |
| Zielgewebe |
Blutgefäße, Bronchien, Uterus, Blase, Darm, Leber, exokrine Drüsen, Iris |
Blutgefäße, Darm, Pankreas, Hirnstamm, Thrombozyten |
Herz Speichel- drüsen |
Blutgefäße, Herz, Bronchien, Uterus, Blase, Darm, Leber, Ziliarmuskel, Mastzellen |
Fettgewebe Skelettmuskel Blasenwand |
| Wirkungen |
Kontraktion: Bronchien, Uterus (Wehen), Blasensphinkter, Urethra (Ejakulation), m. dilatator pupillae Relaxation: Glykogenolyse Salivation Blutgefäße, GI-Trakt |
Blutgefäße↓↑ Präsynaptische Hemmung der Transmitter- freisetzung an sympathischen / parasymp. Endigungen (Darm: Relaxation) Insulinsekretion↓ Plättchen- aggregation |
Herzqualitäten (positiv ino-, chrono-, lusi-, dromo-, bathmotrop) Amylase- sekretion |
Vasodilatation Broncho- dilatation Relaxation Darm Gluco- neogenese Histamin- freisetzung↓ Noradrenalin- freisetzung↑ |
↑Lipolyse in weißem / Thermogenese in braunem Fettgewebe und Skelettmuskel Entspannung des Detrusors (Blase) |
| Kopplung G-Protein |
Gq |
Gi/Go |
Gs |
Gs |
Gs |
| Mechanismus |
PLC + IP3↑ DAG↑ Ca++↑ |
cAMP↓ K+-Kanäle↑ Ca++-Kanäle↓ |
cAMP↑ | cAMP↑ | cAMP↑ |
| Selektive Agonisten |
Phenylephrin |
Clonidin |
Dobutamin |
Salbutamol etc |
Mirabegron |
| (Nicht-) selektive Antagonisten |
Prazosin (Phentolamin) |
Yohimbin (Phentolamin) |
Atenolol Metoprolol |
Butoxamin |
- |
| Wirkung Agonisten |
NA > A > Iso |
A > NA >> Iso |
Iso > NA > A |
Iso > A > NA |
Iso > NA = A |
Die Rezeptorverteilung wird therapeutisch als Ansatzpunkt für selektive Wirkungen genutzt: Beispielsweise α1-Agonisten gegen verstopfte Nasenwege, α2-Antagonisten gegen Impotenz, ß1-Agonisten zur Herzstärkung, ß2-Agonisten als Bronchienerweiterer.| ß-Adrenozeptoren aktivieren über Gs-Proteine die Adenylylcyclase, [cAMP] steigt an. Aktivierte ß1-Adrenozeptoren öffnen L-Typ-Ca++-Kanäle und wirken positiv inotrop |
 Abbildung: Adrenozeptoren und Wirkungen
Abbildung: Adrenozeptoren und Wirkungen α1-Rezeptoren vermitteln alle sympathischen Effekte, die auf
Kontraktion glatter Muskulatur beruhen (Blutgefäße, Blasenausgang,
Uterus / Samenleiter, dilatator pupillae) Speichelsekretion Glykogenolyse in der Leber α2-Rezeptoren vermitteln die präsynaptische Selbsthemmung der
Noradrenalinfreisetzung hemmen die Freisetzung einiger Transmitter und
von Insulin fördern Plättchenaggregation und Vasokonstriktion ß1-Rezeptoren wirken am Herzmuskel positiv inotrop, chronotrop, dromotrop ("Herzqualitäten"); ß1Rezeptorblocker dämpfen die Herztätigkeit. ß2-Rezeptoren vermitteln die Dilatation glatter Muskulatur
in Luftwegen, Koronararterien und Skelettmuskelarteriolen,
sowie in Darm, detrusor vesicae, Uterus, Samenleiter, Ziliarmuskel regen Insulinausschüttung an ß-Zellen (Inselzellen im Pankreas) an, fördern Glykogenolyse und Glukoneogenese in der Leber ß3-Rezeptoren vermitteln Lipolyse im Fettgewebe sowie Thermogenese im Skelettmuskel über Anregung von α1- und α2-Rezeptoren in der Leber vorübergehende Hyperkaliämie, indem sie calciumabhängige Kaliumkanäle öffnen (Kaliumausstrom aus den Zellen). Die Stimulierung von ß1-und ß2-Rezeptoren im Muskelgewebe zeitigt über Anregung der Na/K-Pumpe eine darauffolgende Hypokaliämie (bedingt durch Kaliumaufnahme in die Zellen). Serotonin
α1-Rezeptoren vermitteln alle sympathischen Effekte, die auf
Kontraktion glatter Muskulatur beruhen (Blutgefäße, Blasenausgang,
Uterus / Samenleiter, dilatator pupillae) Speichelsekretion Glykogenolyse in der Leber α2-Rezeptoren vermitteln die präsynaptische Selbsthemmung der
Noradrenalinfreisetzung hemmen die Freisetzung einiger Transmitter und
von Insulin fördern Plättchenaggregation und Vasokonstriktion ß1-Rezeptoren wirken am Herzmuskel positiv inotrop, chronotrop, dromotrop ("Herzqualitäten"); ß1Rezeptorblocker dämpfen die Herztätigkeit. ß2-Rezeptoren vermitteln die Dilatation glatter Muskulatur
in Luftwegen, Koronararterien und Skelettmuskelarteriolen,
sowie in Darm, detrusor vesicae, Uterus, Samenleiter, Ziliarmuskel regen Insulinausschüttung an ß-Zellen (Inselzellen im Pankreas) an, fördern Glykogenolyse und Glukoneogenese in der Leber ß3-Rezeptoren vermitteln Lipolyse im Fettgewebe sowie Thermogenese im Skelettmuskel über Anregung von α1- und α2-Rezeptoren in der Leber vorübergehende Hyperkaliämie, indem sie calciumabhängige Kaliumkanäle öffnen (Kaliumausstrom aus den Zellen). Die Stimulierung von ß1-und ß2-Rezeptoren im Muskelgewebe zeitigt über Anregung der Na/K-Pumpe eine darauffolgende Hypokaliämie (bedingt durch Kaliumaufnahme in die Zellen). Serotonin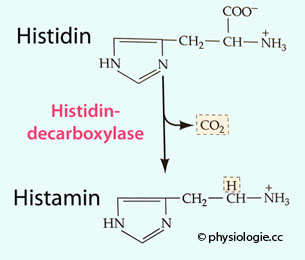 Abbildung: Histaminsynthese Histamin entsteht in mehreren Geweben durch Einwirken des induzierbaren Enzyms Histidin-Decarboxylase auf die essentielle Aminosäure Histidin (Abbildung).
Histamin wird nach seiner Freisetzung in benachbarte Zellen aufgenommen und intrazellulär metabolisiert: Histamin-Methyltransferase (HNMT) findet sich vorwiegend in der Colonschleimhaut, Leber, Milz, und Lunge; Diaminooxidase (DAO) kommt ubiquitär vor, sie baut u.a. Histamin ab (Histaminase), das mit der Nahrung in den Körper gelangt ist. Abbildung) die jeweilige biologische Reaktion. Hohe Konzentrationswerte finden sich in
Abbildung: Histaminsynthese Histamin entsteht in mehreren Geweben durch Einwirken des induzierbaren Enzyms Histidin-Decarboxylase auf die essentielle Aminosäure Histidin (Abbildung).
Histamin wird nach seiner Freisetzung in benachbarte Zellen aufgenommen und intrazellulär metabolisiert: Histamin-Methyltransferase (HNMT) findet sich vorwiegend in der Colonschleimhaut, Leber, Milz, und Lunge; Diaminooxidase (DAO) kommt ubiquitär vor, sie baut u.a. Histamin ab (Histaminase), das mit der Nahrung in den Körper gelangt ist. Abbildung) die jeweilige biologische Reaktion. Hohe Konzentrationswerte finden sich in Haut,
Bronchialschleimhaut, Darmmukosa.
Im Gehirn findet
sich Histamin in etwa gleichen Mengen in Mastzellen und in histaminergen Neuronen (die Zellkörper befinden sich vor allem im Hypothalamus, die Neuriten projizieren in weite
Teile des ZNS (Kortex, Thalamus, Hirnstammkerne) und beteiligen sich an Weckreaktionen, Aufmerksamkeit und der Steuerung des Schlaf-Wach-Rhythmus).
Haut,
Bronchialschleimhaut, Darmmukosa.
Im Gehirn findet
sich Histamin in etwa gleichen Mengen in Mastzellen und in histaminergen Neuronen (die Zellkörper befinden sich vor allem im Hypothalamus, die Neuriten projizieren in weite
Teile des ZNS (Kortex, Thalamus, Hirnstammkerne) und beteiligen sich an Weckreaktionen, Aufmerksamkeit und der Steuerung des Schlaf-Wach-Rhythmus). Im anaphylaktischen Schock kann die Plasma-Histaminkonzentration mehr als 100-fach ansteigen.
Im anaphylaktischen Schock kann die Plasma-Histaminkonzentration mehr als 100-fach ansteigen. Abbildung: Histaminrezeptoren H1-Rezeptoren finden sich im Gehirn, wirken auf Kreislauf und spielen bei allergischen Entzündungen eine Rolle H2-Rezeptoren
sind essentiell für die Sekretion von Magensäure, finden sich aber auch
in glatter Muskulatur und im Gehirn (Großhirnrinde, Basalganglien,
limbisches System) H3-Rezeptoren
beteiligen sich an der Übermittlung neuronaler Impulse (Gehirnrinde,
Thalamus, Hypothalamus) und wirken u.a. auf Bewegungskontrolle
(Basalganglien) und Bewusstsein H4-Rezeptoren modifizieren Immunreaktionen, finden sich auch im Vestibularsystem u.a. Abbildung), genannt H1 bis H4. Diese wirken über verschiedene
G-Proteine: H1 über Gq/11-Protein, aktivieren Phospholipase C, steigern [Ca++] im Zytoplasma, entspannen einige Gefäße (indirekt über NO: Histamininjektion führt - bei intaktem Endothel - zu Blutdruckerniedrigung),
können aber auch (direkte Wirkung) Gefäße, Bronchien, Darm,
Uterusmuskulatur kontrahieren. Weiters führen sie zu Freisetzung von Katecholaminen im Nebennierenmark; Anregung der
Östrogensynthese von Granulosazellen im Ovar; Weckreaktion; Erbrechen H2 über Gs-Protein, aktivieren Adenylylcyclase (steigern cAMP). Sie regen die Säure- und Pepsinproduktion im Magen an (H2-Rezeptorblocker bei ulcus pepticum!), wirken gefäßrelaxierend und positiv ino- / chronotrop am Herzen H3 über Gi/o-Protein,
hemmen Adenylylcyclase (senken cAMP) und wirken über MAP-Kinase und den
Akt-PI3K-Mechanismus (Akt = Protein Kinase B, PI3K =
Phosphatidylinositol 3-Kinase). Sie dämpfen die Neurotransmitter- (Noradrenalin-) Freisetzung im
Gehirn H4 über Gi/o-Protein, hemmen Adenylylcyclase (senken cAMP) - verlagern das Funktionsgleichgewicht von TH1- zu TH2-Zellen, unterstützen Chemotaxis von Mastzellen und Eosinophilen
Abbildung: Histaminrezeptoren H1-Rezeptoren finden sich im Gehirn, wirken auf Kreislauf und spielen bei allergischen Entzündungen eine Rolle H2-Rezeptoren
sind essentiell für die Sekretion von Magensäure, finden sich aber auch
in glatter Muskulatur und im Gehirn (Großhirnrinde, Basalganglien,
limbisches System) H3-Rezeptoren
beteiligen sich an der Übermittlung neuronaler Impulse (Gehirnrinde,
Thalamus, Hypothalamus) und wirken u.a. auf Bewegungskontrolle
(Basalganglien) und Bewusstsein H4-Rezeptoren modifizieren Immunreaktionen, finden sich auch im Vestibularsystem u.a. Abbildung), genannt H1 bis H4. Diese wirken über verschiedene
G-Proteine: H1 über Gq/11-Protein, aktivieren Phospholipase C, steigern [Ca++] im Zytoplasma, entspannen einige Gefäße (indirekt über NO: Histamininjektion führt - bei intaktem Endothel - zu Blutdruckerniedrigung),
können aber auch (direkte Wirkung) Gefäße, Bronchien, Darm,
Uterusmuskulatur kontrahieren. Weiters führen sie zu Freisetzung von Katecholaminen im Nebennierenmark; Anregung der
Östrogensynthese von Granulosazellen im Ovar; Weckreaktion; Erbrechen H2 über Gs-Protein, aktivieren Adenylylcyclase (steigern cAMP). Sie regen die Säure- und Pepsinproduktion im Magen an (H2-Rezeptorblocker bei ulcus pepticum!), wirken gefäßrelaxierend und positiv ino- / chronotrop am Herzen H3 über Gi/o-Protein,
hemmen Adenylylcyclase (senken cAMP) und wirken über MAP-Kinase und den
Akt-PI3K-Mechanismus (Akt = Protein Kinase B, PI3K =
Phosphatidylinositol 3-Kinase). Sie dämpfen die Neurotransmitter- (Noradrenalin-) Freisetzung im
Gehirn H4 über Gi/o-Protein, hemmen Adenylylcyclase (senken cAMP) - verlagern das Funktionsgleichgewicht von TH1- zu TH2-Zellen, unterstützen Chemotaxis von Mastzellen und Eosinophilen Histamin agiert im Nervensystem als Neuromodulator:
Sie beteiligen sich an höheren Funktionen wie Schlaf-Wach-Regulation,
Lernen und Gedächtnis, Immunität, Nahrungs- und Wasseraufnahme oder
Temperaturregulation Im Magen regt es die Salzsäureproduktion und Pepsinogenfreisetzung an (H2-Rezeptoren), im Darm stimuliert es die Sekretion von Kalium- und Chloridionen und fördert über H1-Rezeptoren die Peristaltik im Ileum In den Bronchien kann es rasch adaptierende Dehnungsrezeptoren reizen und wirkt über H1-Rezeptoren bronchienverengend Auf den Uterus wirkt es wehenfördernd (über H1-Rezeptoren) An der Haut: Subkutane Injektion von Histamin führt zur triple response, hauptsächlich über H1-Rezeptoren bewirkt: 1) Sofortige punktförmige Rötung an der Einstichstelle (Vasodilatation von Arteriolen / präkapillären Sphinkteren) 2) flüchtiges Erythem (flush) um die Einstichstelle nach ca. einer halben Minute (Vasodilatation durch einen Axonreflex, vermittelt über Stoffe wie CGRP) 3) Quaddelbildung (Permeabilitätssteigerung) und Juckreiz Im Gewebe ist es bei der Entstehung von Schmerz und Entzündungsprozessen beteiligt. Bei Allergien wirkt es insbesondere im Sinne einer Typ-I-Reaktion (Mastzellendegranulation) Im Herzen wirkt Histamin über H2-Rezeptoren positiv chronotrop und steigert das Herzzeitvolumen Es wirkt allgemein gefäßerweiternd und steigert über H1-Rezeptoren die Permeabilität
postkapillärer Venolen (elektronenmikroskopisch darstellbare Verbreiterung der Spalten
zwischen Endothelzellen, vermutlich bedingt durch
Endothelzell-Kontraktion) Antihistaminika lindern histaminbedingte allergische Symptome, indem sie H1- und H2-GPCRs blockieren. Einige wirken auch als Antiemetika und werden gegen Kinetosen ("Reisekrankheit") eingesetzt.
Histamin agiert im Nervensystem als Neuromodulator:
Sie beteiligen sich an höheren Funktionen wie Schlaf-Wach-Regulation,
Lernen und Gedächtnis, Immunität, Nahrungs- und Wasseraufnahme oder
Temperaturregulation Im Magen regt es die Salzsäureproduktion und Pepsinogenfreisetzung an (H2-Rezeptoren), im Darm stimuliert es die Sekretion von Kalium- und Chloridionen und fördert über H1-Rezeptoren die Peristaltik im Ileum In den Bronchien kann es rasch adaptierende Dehnungsrezeptoren reizen und wirkt über H1-Rezeptoren bronchienverengend Auf den Uterus wirkt es wehenfördernd (über H1-Rezeptoren) An der Haut: Subkutane Injektion von Histamin führt zur triple response, hauptsächlich über H1-Rezeptoren bewirkt: 1) Sofortige punktförmige Rötung an der Einstichstelle (Vasodilatation von Arteriolen / präkapillären Sphinkteren) 2) flüchtiges Erythem (flush) um die Einstichstelle nach ca. einer halben Minute (Vasodilatation durch einen Axonreflex, vermittelt über Stoffe wie CGRP) 3) Quaddelbildung (Permeabilitätssteigerung) und Juckreiz Im Gewebe ist es bei der Entstehung von Schmerz und Entzündungsprozessen beteiligt. Bei Allergien wirkt es insbesondere im Sinne einer Typ-I-Reaktion (Mastzellendegranulation) Im Herzen wirkt Histamin über H2-Rezeptoren positiv chronotrop und steigert das Herzzeitvolumen Es wirkt allgemein gefäßerweiternd und steigert über H1-Rezeptoren die Permeabilität
postkapillärer Venolen (elektronenmikroskopisch darstellbare Verbreiterung der Spalten
zwischen Endothelzellen, vermutlich bedingt durch
Endothelzell-Kontraktion) Antihistaminika lindern histaminbedingte allergische Symptome, indem sie H1- und H2-GPCRs blockieren. Einige wirken auch als Antiemetika und werden gegen Kinetosen ("Reisekrankheit") eingesetzt.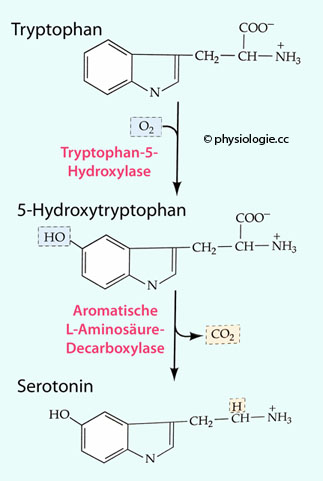 Abbildung: Serotoninsynthese
Serotonin wird zwar mit der Nahrung zugeführt, erreicht aber auf diesem Weg den
Kreislauf kaum, da es schon vorher metabolisiert wird. Chromaffine Zellen
und serotoninproduzierende Neuronen bilden es aus der essentiellen Aminosäure Tryptophan über 5-OH-Tryptophan neu (Abbildung). Serotonintransporter (SERT) sind wichtige Angriffspunkte für psychoaktive Pharmaka, insbesondere Antidepressiva, Anxiolytika und Stimulantien (vgl. dort). Selektive Wiederaufnahmehemmer (SSRIs, selective serotonin reuptake inhibitors) werden als Antidepressiva (z.B. Prozac) verwendet. Nicht recyceltes Serotonin wird mittels Monoaminooxidase (MAO) und Aldehyd-Dehydrogenase abgebaut (hauptsächlich decarboxyliert). Der Hauptmetabolit des Sertonoinabbaus, 5-Hydroxyindolessigsäure (5-HIAA, 5-hydroxyindoleacetic acid) gelangt in Kreislauf, Niere und Harn. Die HIAA-Ausscheidung mit dem Urin ist ein Maß für die Serotoninproduktion des Körpers. in enterochromaffinähnlichen (ECL: Enterochromaffin-like)
Zellen des Darms (mehr als 90% des Serotonin- Körperbestandes). Denen
im Nebennierenmark ähnelnde Zellen sind in der intestinalen Mukosa
zwischen Enterozyten eingelagert und finden sich vorwiegend
in Magen und Dünndarm. Einige Neuronen des plexus myentericus enthalten
ebenfalls 5-HT, das hier als exzitatorischer Transmitter wirkt; in Speichergranula von Blutplättchen.
Diese reichern 5-HT via aktiven Transport aus dem Blutplasma an und
setzen es frei, wenn sie aktiviert werden und aggregieren (das Serum
von geronnenem Blut weist hohe Serotoninkonzentration auf); verteilt im Zentralnervensystem, wo es insbesondere von Neuronen produziert wird, deren Soma sich im Mittelhirn befindet (Raphekerne). Serotonin-Wiederaufnahmehemmer (SSRI: Selective serotonin reuptake inhibitors) erhöhen die Serotoninmenge im
Synapsenspalt, stimulieren präsynaptische Autorezeptoren und veranlassen sie, die
Serotoninproduktion des Neuriten zu reduzieren. Im Verlauf mehrerer
Wochen verlieren die Rezeptoren ihre Empfindlichkeit (Adaptation),
postsynaptische Serotoninrezeptoren können hingegen ihre Sensitivität
sogar steigern.
Abbildung: Serotoninsynthese
Serotonin wird zwar mit der Nahrung zugeführt, erreicht aber auf diesem Weg den
Kreislauf kaum, da es schon vorher metabolisiert wird. Chromaffine Zellen
und serotoninproduzierende Neuronen bilden es aus der essentiellen Aminosäure Tryptophan über 5-OH-Tryptophan neu (Abbildung). Serotonintransporter (SERT) sind wichtige Angriffspunkte für psychoaktive Pharmaka, insbesondere Antidepressiva, Anxiolytika und Stimulantien (vgl. dort). Selektive Wiederaufnahmehemmer (SSRIs, selective serotonin reuptake inhibitors) werden als Antidepressiva (z.B. Prozac) verwendet. Nicht recyceltes Serotonin wird mittels Monoaminooxidase (MAO) und Aldehyd-Dehydrogenase abgebaut (hauptsächlich decarboxyliert). Der Hauptmetabolit des Sertonoinabbaus, 5-Hydroxyindolessigsäure (5-HIAA, 5-hydroxyindoleacetic acid) gelangt in Kreislauf, Niere und Harn. Die HIAA-Ausscheidung mit dem Urin ist ein Maß für die Serotoninproduktion des Körpers. in enterochromaffinähnlichen (ECL: Enterochromaffin-like)
Zellen des Darms (mehr als 90% des Serotonin- Körperbestandes). Denen
im Nebennierenmark ähnelnde Zellen sind in der intestinalen Mukosa
zwischen Enterozyten eingelagert und finden sich vorwiegend
in Magen und Dünndarm. Einige Neuronen des plexus myentericus enthalten
ebenfalls 5-HT, das hier als exzitatorischer Transmitter wirkt; in Speichergranula von Blutplättchen.
Diese reichern 5-HT via aktiven Transport aus dem Blutplasma an und
setzen es frei, wenn sie aktiviert werden und aggregieren (das Serum
von geronnenem Blut weist hohe Serotoninkonzentration auf); verteilt im Zentralnervensystem, wo es insbesondere von Neuronen produziert wird, deren Soma sich im Mittelhirn befindet (Raphekerne). Serotonin-Wiederaufnahmehemmer (SSRI: Selective serotonin reuptake inhibitors) erhöhen die Serotoninmenge im
Synapsenspalt, stimulieren präsynaptische Autorezeptoren und veranlassen sie, die
Serotoninproduktion des Neuriten zu reduzieren. Im Verlauf mehrerer
Wochen verlieren die Rezeptoren ihre Empfindlichkeit (Adaptation),
postsynaptische Serotoninrezeptoren können hingegen ihre Sensitivität
sogar steigern. Abbildung: Metabotroper Serotoninrezeptor ERK 1/2: Extracellular signal-regulated kinases 1/2; klassische MAP-Kinasen
mTOR: mechanistic target of rapamycin, phosphatübertragendes Enzym in der Signaltransduktionskette, wichtig für Zellwachstum und -Mobilität 5-HT1-Rezeptoren (5 Arten: 5-HT1A, 5-HT1B, 5-HT1D, 5-HT1E, 5-HT1F) koppeln an Gi/G0, hemmen die Adenylylcyclase und damit die Bildung von cAMP in der Zielzelle. Sie senken den Blutdruck, wirken anxiolytisch (5-HT1A-Rezeptoren haben starken Einfluß auf Gemütslage und Verhalten), kontrahieren Blutgefäße in Meningen und Koronarien (5-HT1B: Migräne!) und wirken im Trigeminusgebiet schmerzhemmend (5-HT1D). 5-HT1A und 5-HT1B erhöhen die Kaliumleitfähigkeit und hyperpolarisieren die Zelle, wirken also inhibitorisch; 5-HT2-Rezeptoren (3 Subtypen: 5-HT2A, 5-HT2B, 5-HT2C) vermitteln ihre Effekte über Gq/G11-Proteine und steigern IP3 und zytoplasmatisches [Ca++].
Ihre physiologische Bedeutung ist vermutlich limitiert, bei
pathologischen Zuständen kann sie groß sein (z.B. Thrombosen, Asthma).
Sie kommen im Gehirn sowie peripher vor (vasoaktiv, Blutplättchen,
autonome Nerven). Einige kontrahieren Blutgefäße und gastrointestinale Muskulatur,
aktivieren Thrombozyten und haben psychotrope Wirkung (5-HT2A); andere setzen NO aus Endothelien frei (5-HT2B) oder wirken über den Hypothalamus appetitzügelnd (5-HT2C). 5-HT2A und 5-HT2C senken die Kaliumleitfähigkeit und depolarisieren die Zelle, wirken also exzitatorisch; 5-HT3-Rezeptoren (bzw. 5-HT3A-, M-Rezeptoren) sind ligandengesteuerte Na+-K+-Permeasen (kein second messenger- Mechanismus notwendig) und finden sich auf Nervenzellen, die sie rasch depolarisieren. Man findet sie im zentralen peripheren Nervensystem, insbesondere an nozizeptiven und enterischen Fasern. Sie sind am Bezold-Jarisch-Reflex und am Brechreflex beteiligt (Rezeptoren in area postrema und nucl. tractus solitarii). Sie regen ferner die Freisetzung von CCK im ZNS sowie von Acetylcholin im Gastrointestinaltrakt an; 5-HT4-Rezeptoren sind GS-Protein-gekoppelt
und wirken über Steigerung der [cAMP]. Sie kommen im peripheren wie im
zentralen Nervensystem vor und wirken exzitatorisch. Sie agieren im Darm peristaltikfördernd und am Herzen positiv inotrop und chronotrop, senken die Kaliumleitfähigkeit und depolarisieren (langsam) die Zelle; 5-HT5A-Rezeptoren (5-HT5B bei Mäusen) sind ebenfalls GS-Protein-gekoppelt und wirken über Steigerung der [cAMP]. 5-HT5-Rezeptoren befinden sich im ZNS, bei Nagetieren ändern sie das Verhalten, die Wirkungen beim Menschen sind nicht klar; 5-HT6-Rezeptoren - GS-Protein, ↑[cAMP] - auf Nervenzellen (Beeinflussung der Neurotransmission) und Leukozyten, die Wirkungen beim Menschen sind nicht klar; 5-HT7-Rezeptoren ( Abbildung) - GS/G12-Protein, ↑[cAMP]
finden sich im ZNS und im Gastrointestinaltrakt. Wahrscheinlich sind
sie beteiligt an zirkadianen Rhythmen, Thermoregulation u.a.,
vielleicht auch an der Entstehung von Migräne. Peptidhormone bzw. -mediatoren sind z.B. TRH (3 AS), Bradykinin (9 AS), Angiotensin II (10 AS), GnRH (10 AS), Vasopressin (11 AS), Oxytocin (11 AS), Somatostatin (14 AS), ANP (28 AS), ACTH (39 AS), CRH (41 AS), GHRH (43 AS). Zahlreiche
Peptide wirken sowohl als Hormone als auch als Neuropeptide:
Nervenzellen, die sie bilden, setzen sie bei höherer
Aktionspotentialfrequenz frei. Neuropeptide wirken als "klassische" Transmitter; zu ihnen zählen u.a. VIP, NPY, Substanz P, Opioide. Neuropeptide sind klein- bis mittelmolekular (sie bestehen aus einigen wenigen bis zu mehreren Dutzend Aminosäuren), mehr als 100 sind bekannt sind. Sie wirken über G-Proteine, z.T. auch als Hormone (z.B. Insulin, Somatostatin, Cholecystokinin, Serotonin, GLP-1). Sie finden sich im Nervensystem und in zahlreichen peripheren Geweben und fungieren oft als Cotransmitter,
d.h. sie werden zusammen mit einem Nichtpeptid (z.B. Acetylcholin,
Noradrenalin) freigesetzt, dessen Wirkung sie ergänzen bzw. erweitern.
Abbildung: Metabotroper Serotoninrezeptor ERK 1/2: Extracellular signal-regulated kinases 1/2; klassische MAP-Kinasen
mTOR: mechanistic target of rapamycin, phosphatübertragendes Enzym in der Signaltransduktionskette, wichtig für Zellwachstum und -Mobilität 5-HT1-Rezeptoren (5 Arten: 5-HT1A, 5-HT1B, 5-HT1D, 5-HT1E, 5-HT1F) koppeln an Gi/G0, hemmen die Adenylylcyclase und damit die Bildung von cAMP in der Zielzelle. Sie senken den Blutdruck, wirken anxiolytisch (5-HT1A-Rezeptoren haben starken Einfluß auf Gemütslage und Verhalten), kontrahieren Blutgefäße in Meningen und Koronarien (5-HT1B: Migräne!) und wirken im Trigeminusgebiet schmerzhemmend (5-HT1D). 5-HT1A und 5-HT1B erhöhen die Kaliumleitfähigkeit und hyperpolarisieren die Zelle, wirken also inhibitorisch; 5-HT2-Rezeptoren (3 Subtypen: 5-HT2A, 5-HT2B, 5-HT2C) vermitteln ihre Effekte über Gq/G11-Proteine und steigern IP3 und zytoplasmatisches [Ca++].
Ihre physiologische Bedeutung ist vermutlich limitiert, bei
pathologischen Zuständen kann sie groß sein (z.B. Thrombosen, Asthma).
Sie kommen im Gehirn sowie peripher vor (vasoaktiv, Blutplättchen,
autonome Nerven). Einige kontrahieren Blutgefäße und gastrointestinale Muskulatur,
aktivieren Thrombozyten und haben psychotrope Wirkung (5-HT2A); andere setzen NO aus Endothelien frei (5-HT2B) oder wirken über den Hypothalamus appetitzügelnd (5-HT2C). 5-HT2A und 5-HT2C senken die Kaliumleitfähigkeit und depolarisieren die Zelle, wirken also exzitatorisch; 5-HT3-Rezeptoren (bzw. 5-HT3A-, M-Rezeptoren) sind ligandengesteuerte Na+-K+-Permeasen (kein second messenger- Mechanismus notwendig) und finden sich auf Nervenzellen, die sie rasch depolarisieren. Man findet sie im zentralen peripheren Nervensystem, insbesondere an nozizeptiven und enterischen Fasern. Sie sind am Bezold-Jarisch-Reflex und am Brechreflex beteiligt (Rezeptoren in area postrema und nucl. tractus solitarii). Sie regen ferner die Freisetzung von CCK im ZNS sowie von Acetylcholin im Gastrointestinaltrakt an; 5-HT4-Rezeptoren sind GS-Protein-gekoppelt
und wirken über Steigerung der [cAMP]. Sie kommen im peripheren wie im
zentralen Nervensystem vor und wirken exzitatorisch. Sie agieren im Darm peristaltikfördernd und am Herzen positiv inotrop und chronotrop, senken die Kaliumleitfähigkeit und depolarisieren (langsam) die Zelle; 5-HT5A-Rezeptoren (5-HT5B bei Mäusen) sind ebenfalls GS-Protein-gekoppelt und wirken über Steigerung der [cAMP]. 5-HT5-Rezeptoren befinden sich im ZNS, bei Nagetieren ändern sie das Verhalten, die Wirkungen beim Menschen sind nicht klar; 5-HT6-Rezeptoren - GS-Protein, ↑[cAMP] - auf Nervenzellen (Beeinflussung der Neurotransmission) und Leukozyten, die Wirkungen beim Menschen sind nicht klar; 5-HT7-Rezeptoren ( Abbildung) - GS/G12-Protein, ↑[cAMP]
finden sich im ZNS und im Gastrointestinaltrakt. Wahrscheinlich sind
sie beteiligt an zirkadianen Rhythmen, Thermoregulation u.a.,
vielleicht auch an der Entstehung von Migräne. Peptidhormone bzw. -mediatoren sind z.B. TRH (3 AS), Bradykinin (9 AS), Angiotensin II (10 AS), GnRH (10 AS), Vasopressin (11 AS), Oxytocin (11 AS), Somatostatin (14 AS), ANP (28 AS), ACTH (39 AS), CRH (41 AS), GHRH (43 AS). Zahlreiche
Peptide wirken sowohl als Hormone als auch als Neuropeptide:
Nervenzellen, die sie bilden, setzen sie bei höherer
Aktionspotentialfrequenz frei. Neuropeptide wirken als "klassische" Transmitter; zu ihnen zählen u.a. VIP, NPY, Substanz P, Opioide. Neuropeptide sind klein- bis mittelmolekular (sie bestehen aus einigen wenigen bis zu mehreren Dutzend Aminosäuren), mehr als 100 sind bekannt sind. Sie wirken über G-Proteine, z.T. auch als Hormone (z.B. Insulin, Somatostatin, Cholecystokinin, Serotonin, GLP-1). Sie finden sich im Nervensystem und in zahlreichen peripheren Geweben und fungieren oft als Cotransmitter,
d.h. sie werden zusammen mit einem Nichtpeptid (z.B. Acetylcholin,
Noradrenalin) freigesetzt, dessen Wirkung sie ergänzen bzw. erweitern.
| Melanocortinrezeptoren: Verteilung und Wirkungen Modifiziert nach Elmér F, Kotaleski JH, Lansner A. Melanocortin-Receptor Modelling (2006) |
||
| Rezeptor |
Vorkommen |
Hauptwirkung(en) |
| MC1R (auch Melanotropinrezeptor, MSHR etc) | Melanozyten, Makrophagen / Immunzellen, Astrozyten, Epithelzellen, Fibroblasten u.a. | Melanogenese (Pigmentierung), Entzündungshemmung, Schmerzkontrolle |
| MC2R Klassischer ACTH-Rezeptor |
Nebennierenrinde, Fettzellen | Steroidsynthese |
| MC3R Bindet MSH und ACTH |
Gehirn, Gastrointestinaltrakt, Plazenta, Herzmuskel Makrophagen |
Energiehaushalt, Entzündungshemmung |
| MC4R Bindet MSH und ACTH |
Gehirn | Energiehaushalt Erektion |
| MC5R | Haut (exokrine Drüsen), Nebennieren, Lymphozyten |
Talgproduktion, Erythrozytenentwicklung, Thermoregulation |
Neurokinine
Über NK1-Rezeptoren erfolgen Vasodilatation und
transvaskuläre Flüssigkeitsfiltration, Chemotaxis von Leukozyten,
erhöhte Sensitivität spinaler Schmerzneuronen. Freisetzung
von Histamin und Prostaglandinen aus Mastzellen (neurogene Entzündung)
verursacht so einerseits Schmerz, trägt andererseits zur Wundheilung
bei
Über NK2-Rezeptoren wird der Darm kontrahiert
Über NK3-Rezeptoren erfolgt Freisetzung von Acetylcholin an cholinergen Nervenfasern. Substanz P s. dort NPY s. dort Die Synthese erfolgt in neuroendokrinen / parasympathischen Zellen sowie in D-Zellen des Pankreas. VIP wird von Hepatozyten inaktiviert.Verteilung von Endothelinen / Endothelinrezeptoren Nach Ritter / Flower / Henderson / Loke / MacEwan / Rang, Rang & Dale's Pharmacology, 9th ed. Elsevier 2020 |
|||||
| Gewebe |
Endothelin |
Rezeptor |
|||
| |
1 |
2 |
3 |
ETA |
ETB |
| Endothel |
++++ |
- |
- |
+ |
|
| Glatter Muskel |
+ |
- |
- |
++ |
- |
| Gehirn |
+++ |
+ |
+ |
+++ |
|
| Nieren |
++ |
++ |
+ |
+ |
++ |
| Darm |
+ |
+ |
+++ |
+ |
+++ |
| Nebenniere |
+ |
- |
+++ |
+ |
++ |
Freisetzung von hypothalamisch-hypophysären Hormonen, Aldosteron, Adrenalin, natriuretischen Peptiden Renale Ausscheidung von Wasser und Kochsalz Synthese von Thyreoglobulin (hohe ET1-Konzentration im Kolloid der Schilddrüsenfollikel) Steuerung des plazentaren Kreislaufs (hohe ET1-Konzentration in der Amnionflüssigkeit) Gefäßkontraktion (Vasospasmen) in Gehirn und Nieren Entwicklung des kardiorespiratorischen Systems (ET1-Antagonisten wirken teratogen) Abbildung), ihre Gesamtwirkung ist Vasokonstriktion.  Abbildung: Systemische Wirkung der Aktivierung von Endothelinrezeptoren Aktivierung von ETA-Rezeptoren (links) verursacht starke Vasokonstriktion, Blutdruckanstieg und Natriumretention durch Wirkung auf Nervensystem, Herz und Kreislauf, Nieren und Nebennieren. ETA stimuliert auch die Freisetzung von ANP und könnte so die hypertensiven Effekte abmildern.Aktivierung von ETB-Rezeptoren (rechts) verursacht Blutdrucksenkung und Salzausscheidung - über mehrere Wege:
Abbildung: Systemische Wirkung der Aktivierung von Endothelinrezeptoren Aktivierung von ETA-Rezeptoren (links) verursacht starke Vasokonstriktion, Blutdruckanstieg und Natriumretention durch Wirkung auf Nervensystem, Herz und Kreislauf, Nieren und Nebennieren. ETA stimuliert auch die Freisetzung von ANP und könnte so die hypertensiven Effekte abmildern.Aktivierung von ETB-Rezeptoren (rechts) verursacht Blutdrucksenkung und Salzausscheidung - über mehrere Wege:  NO-Produktion im Endothel (wirkt vasodilatierend), reduzierte Reninfreisetzung, Hemmung der Rückresorption von Wasser und Natrium in der Niere, geringere Katecholaminfreisetzung in der Nebenniere (und vielleicht auch negativ inotrope Wirkung auf das Herz)
Insgesamt bewirkt Stimulierung von ETA-Rezeptoren Natriumretention und Blutdruckerhöhung, Stimulierung von ETB-Rezeptoren
hingegen Natriurese und Blutdrucksenkung. Endotheline erreichen diese
Effekte über Einfluss auf Gefäße, Herz, Nebenniere,
Sympathikusaktivität und vielleicht auch Barorezeptorempfindlichkeit.
ET-1 liegt unter physiologischen Bedingungen in sehr niedriger
Konzentration vor und spielt eine eher geringe Rolle; bei geschädigtem
Endothel aber sind seine Effekte deutlich ausgeprägt.GALR1 in ZNS, Dünndarm, Herz (hemmt die Adelylylcyclase), GALR2 in ZNS und gastrointestinalem System (wirkt über PLC / PKC), GALR3 in zahlreichen Geweben. Stickstoffmonoxid (NO) als kleinstes endogenes Signalmolekül wird durch NO-Synthasen
(NOS) gebildet, wobei Stickstoff aus der Aminosäure L-Arginin
und molekularer Sauerstoff aus dem Blut bezogen
wird. NO ist ein zentrales Signalmolekül im Kreislauf und im
Nervensystem. Es ist fettlöslich und so kurzlebig (Halbwertszeit einige
Sekunden), dass es nur in einem engen Umkreis seiner Entstehung wirksam
sein kann, dabei aber nicht auf eine bestimmte Zielzelle limitiert ist,
sondern frei durch das Gewebe diffundiert.
Das Radikal NO regiert im Körper innerhalb einer halben
Minute mit Sauerstoff und Wasser zu Nitrat und Nitrit; mit dem
Superoxidradikal O2-.
in Sekundenbruchteilen zu Peroxynitrit (NO3-): An der Luft ist NO
stabil genug, um z.B. in der Exspirationsluft
nachgewiesen werden zu können - z.B. zur Diagnose von Asthma,
Atemwegsentzündungen (Bronchitis) - NO wird u.a. in den Atemwegen vermehrt bei
entzündlichen Vorgängen gebildet. Abbildung), Thrombozyten (verhindert Aggregation), oder hilft M1-Makrophagen bei der Phagozytose.
NO-Produktion im Endothel (wirkt vasodilatierend), reduzierte Reninfreisetzung, Hemmung der Rückresorption von Wasser und Natrium in der Niere, geringere Katecholaminfreisetzung in der Nebenniere (und vielleicht auch negativ inotrope Wirkung auf das Herz)
Insgesamt bewirkt Stimulierung von ETA-Rezeptoren Natriumretention und Blutdruckerhöhung, Stimulierung von ETB-Rezeptoren
hingegen Natriurese und Blutdrucksenkung. Endotheline erreichen diese
Effekte über Einfluss auf Gefäße, Herz, Nebenniere,
Sympathikusaktivität und vielleicht auch Barorezeptorempfindlichkeit.
ET-1 liegt unter physiologischen Bedingungen in sehr niedriger
Konzentration vor und spielt eine eher geringe Rolle; bei geschädigtem
Endothel aber sind seine Effekte deutlich ausgeprägt.GALR1 in ZNS, Dünndarm, Herz (hemmt die Adelylylcyclase), GALR2 in ZNS und gastrointestinalem System (wirkt über PLC / PKC), GALR3 in zahlreichen Geweben. Stickstoffmonoxid (NO) als kleinstes endogenes Signalmolekül wird durch NO-Synthasen
(NOS) gebildet, wobei Stickstoff aus der Aminosäure L-Arginin
und molekularer Sauerstoff aus dem Blut bezogen
wird. NO ist ein zentrales Signalmolekül im Kreislauf und im
Nervensystem. Es ist fettlöslich und so kurzlebig (Halbwertszeit einige
Sekunden), dass es nur in einem engen Umkreis seiner Entstehung wirksam
sein kann, dabei aber nicht auf eine bestimmte Zielzelle limitiert ist,
sondern frei durch das Gewebe diffundiert.
Das Radikal NO regiert im Körper innerhalb einer halben
Minute mit Sauerstoff und Wasser zu Nitrat und Nitrit; mit dem
Superoxidradikal O2-.
in Sekundenbruchteilen zu Peroxynitrit (NO3-): An der Luft ist NO
stabil genug, um z.B. in der Exspirationsluft
nachgewiesen werden zu können - z.B. zur Diagnose von Asthma,
Atemwegsentzündungen (Bronchitis) - NO wird u.a. in den Atemwegen vermehrt bei
entzündlichen Vorgängen gebildet. Abbildung), Thrombozyten (verhindert Aggregation), oder hilft M1-Makrophagen bei der Phagozytose.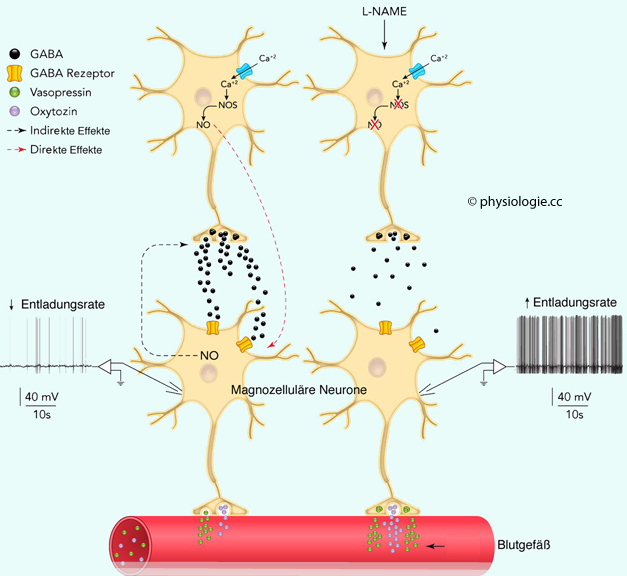 Abbildung: Stickstoffmonoxid (NO) hemmt die Hormonfreisetzung magnozellulärer Neuronen
Das Arginin-Analog Nω-Nitro-L-arginine methyl ester hydrochloride (L-NAME) ist ein membrangängiger NOS-Hemmer - es senkt die Bildung von NO und wirkt u.a. blutdrucksteigernd. NOS1 ist
in ~2% aller Neurone im Gehirn nachweisbar, vor allem in Hypothalamus,
Kleinhirn und Hippocampus, aber auch Großhirnrinde, bulbus olfactorius,
Amygdala, substantia nigra. Makrophagen, neutrophile Granulozyten, Kupffer-Zellen, Endothelzellen, glatte Muskelzellen, Fibroblasten exprimieren NOS2 bei immunologischer Anregung (Kontakt mit Mikroorganismen). NOS3 befindet sich auf Thrombozyten, Endothel- und anderen Zellen.
Anregung von Rezeptoren (für Acetylcholin, Bradykinin, Substanz P usw.) steigert die intrazelluläre [Ca++], Calciumionen lagern sich an Calmodulin, und der Ca++-Calmodulin-Komplex aktiviert NOS. Die Gesamtwirkung hängt von der Gewichtung involvierter Faktoren ab (z.B.: Proteinkinase A erhöht, Proteinkinase C reduziert die NOS3-Aktivität). Mechanische Reizung (Scherkräfte, shear stress) aktiviert die NOS (eNOS bei Endothelzellen) über Serin-Threonin Proteinkinasen (Proteinkinase B - PKB). Proteinkinase B (PKB, auch Akt genannt) ist die Bezeichnung einer Gruppe von Serin/Threonin-Kinasen (drei Isoformen), die bei zahlreichen Vorgängen (Zellwachstum, Transkription, Migration, Glucosestoffwechsel, Apoptose) eine Rolle spielen.
Abbildung: Stickstoffmonoxid (NO) hemmt die Hormonfreisetzung magnozellulärer Neuronen
Das Arginin-Analog Nω-Nitro-L-arginine methyl ester hydrochloride (L-NAME) ist ein membrangängiger NOS-Hemmer - es senkt die Bildung von NO und wirkt u.a. blutdrucksteigernd. NOS1 ist
in ~2% aller Neurone im Gehirn nachweisbar, vor allem in Hypothalamus,
Kleinhirn und Hippocampus, aber auch Großhirnrinde, bulbus olfactorius,
Amygdala, substantia nigra. Makrophagen, neutrophile Granulozyten, Kupffer-Zellen, Endothelzellen, glatte Muskelzellen, Fibroblasten exprimieren NOS2 bei immunologischer Anregung (Kontakt mit Mikroorganismen). NOS3 befindet sich auf Thrombozyten, Endothel- und anderen Zellen.
Anregung von Rezeptoren (für Acetylcholin, Bradykinin, Substanz P usw.) steigert die intrazelluläre [Ca++], Calciumionen lagern sich an Calmodulin, und der Ca++-Calmodulin-Komplex aktiviert NOS. Die Gesamtwirkung hängt von der Gewichtung involvierter Faktoren ab (z.B.: Proteinkinase A erhöht, Proteinkinase C reduziert die NOS3-Aktivität). Mechanische Reizung (Scherkräfte, shear stress) aktiviert die NOS (eNOS bei Endothelzellen) über Serin-Threonin Proteinkinasen (Proteinkinase B - PKB). Proteinkinase B (PKB, auch Akt genannt) ist die Bezeichnung einer Gruppe von Serin/Threonin-Kinasen (drei Isoformen), die bei zahlreichen Vorgängen (Zellwachstum, Transkription, Migration, Glucosestoffwechsel, Apoptose) eine Rolle spielen. 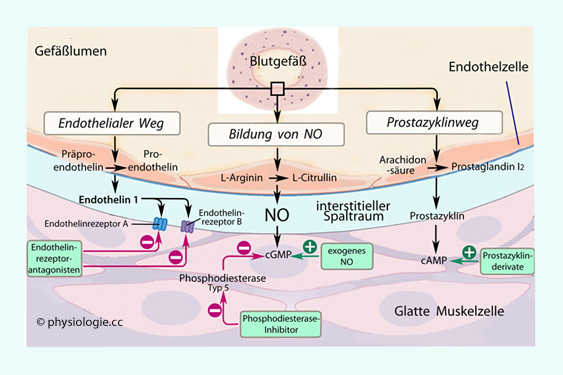 Abbildung: NO, Endothelin und Prostazyklin an der glatten Muskulatur Vasodilatation ( Abbildung) und Blutdrucksenkung (in Widerstandsgefäßen dauernd aktiv, vermutlich auch an
der allgemeinen Vasodilatation der Schwangerschaft beteiligt); im
Herzen bewirken parasympathische (cholinerge)
Fasern NO-Freisetzung, Erweiterung der Koronargefäße und verbesserte
myokardiale Perfusion. NO wirkt auch angiogenetisch (Neubildung von
Blutgefäßen) Hemmung der Plättchenaktivierung, -aggregation und -adhäsion; Verminderung der Expression von Adhäsionsmolekülen Unterdrückung der Transkription leukozytenbindender Adhäsionsmoleküle, wie VCAM-1 (vascular cell adhesion molecule 1), das die Anlagerung von Lymphozyten, Monozyten, eosinophilen und basophilen Granulozyten an das Endothel vermittelt Zentralnervöse Wirkungen (z.B. Unterstützung der
Langzeitpotenzierung). NO wirkt als NANC prä- wie postsynaptisch sowie auf Gliazellen. Aktivierte NMDA-Rezeptoren
stimulieren die NO-Synthase (in dem einen Prozent der zerebralen
Neurone, die über dieses Enzym verfügen), NO diffundiert an
präsynaptische Apparate (so gut wie alle Neurone sind NO-beeinflusst),
induziert die Bildung von cGMP / PIP2, und das mobilisiert den
Vesikelpool, d.h. die glutamaterge Neurotransmission
(Verstärkungseffekt) Peripher (z.B. Entzündungsmarker in oberen Luftwegen, Magenentleerung, Erektion) Pathologische Reize auf das Immunsystem (bakterielle Lipopolysaccharide, und/oder Zytokine, vor allem γ-Interferon) aktivieren die induzierbare iNOS, die dann eine hohe
NO-Syntheseleistung erlangt. NO dient in diesem Fall der Abwehr
(zytotoxische Effekte an Pathogenen, von Viren bis Parasiten sowie
Tumorzellen).
sind kurzlebige Signalmoleküle, die lokal produziert werden und die mit
kleinen benachbarten Zellgruppen kommunizieren (parakrine Wirkung). Sie können bei
entsprechender Reizung durch Wirkung über angeregte G-Proteine aus der
Zellmembran in das Zytoplasma freigesetzt werden. beteiligen sich an der Regulierung des Zellwachstums, wirken bei
Entzündungsvorgängen (fördernd oder hemmend), tragen zur
Schmerzentstehung bei, sind in den Ablauf von Schwangerschaft und
Geburt involviert, beeinflussen Perfusion und Blutdruckkontrolle. gebildet, die aus der Nahrung aufgenommen oder aus
Linolsäure synthetisiert wurde und in Phospholipiden der Zellmembran
gebunden vorliegt.
Abbildung: NO, Endothelin und Prostazyklin an der glatten Muskulatur Vasodilatation ( Abbildung) und Blutdrucksenkung (in Widerstandsgefäßen dauernd aktiv, vermutlich auch an
der allgemeinen Vasodilatation der Schwangerschaft beteiligt); im
Herzen bewirken parasympathische (cholinerge)
Fasern NO-Freisetzung, Erweiterung der Koronargefäße und verbesserte
myokardiale Perfusion. NO wirkt auch angiogenetisch (Neubildung von
Blutgefäßen) Hemmung der Plättchenaktivierung, -aggregation und -adhäsion; Verminderung der Expression von Adhäsionsmolekülen Unterdrückung der Transkription leukozytenbindender Adhäsionsmoleküle, wie VCAM-1 (vascular cell adhesion molecule 1), das die Anlagerung von Lymphozyten, Monozyten, eosinophilen und basophilen Granulozyten an das Endothel vermittelt Zentralnervöse Wirkungen (z.B. Unterstützung der
Langzeitpotenzierung). NO wirkt als NANC prä- wie postsynaptisch sowie auf Gliazellen. Aktivierte NMDA-Rezeptoren
stimulieren die NO-Synthase (in dem einen Prozent der zerebralen
Neurone, die über dieses Enzym verfügen), NO diffundiert an
präsynaptische Apparate (so gut wie alle Neurone sind NO-beeinflusst),
induziert die Bildung von cGMP / PIP2, und das mobilisiert den
Vesikelpool, d.h. die glutamaterge Neurotransmission
(Verstärkungseffekt) Peripher (z.B. Entzündungsmarker in oberen Luftwegen, Magenentleerung, Erektion) Pathologische Reize auf das Immunsystem (bakterielle Lipopolysaccharide, und/oder Zytokine, vor allem γ-Interferon) aktivieren die induzierbare iNOS, die dann eine hohe
NO-Syntheseleistung erlangt. NO dient in diesem Fall der Abwehr
(zytotoxische Effekte an Pathogenen, von Viren bis Parasiten sowie
Tumorzellen).
sind kurzlebige Signalmoleküle, die lokal produziert werden und die mit
kleinen benachbarten Zellgruppen kommunizieren (parakrine Wirkung). Sie können bei
entsprechender Reizung durch Wirkung über angeregte G-Proteine aus der
Zellmembran in das Zytoplasma freigesetzt werden. beteiligen sich an der Regulierung des Zellwachstums, wirken bei
Entzündungsvorgängen (fördernd oder hemmend), tragen zur
Schmerzentstehung bei, sind in den Ablauf von Schwangerschaft und
Geburt involviert, beeinflussen Perfusion und Blutdruckkontrolle. gebildet, die aus der Nahrung aufgenommen oder aus
Linolsäure synthetisiert wurde und in Phospholipiden der Zellmembran
gebunden vorliegt. Aus dieser Bindung muss die Arachidonsäure freigesetzt werden. Das
zytoplasmatische Enzym Phospholipase A2 (PLA2) ermöglicht das. PLA2 wird durch zahlreiche Faktoren reguliert, z.B. wird sie
Aus dieser Bindung muss die Arachidonsäure freigesetzt werden. Das
zytoplasmatische Enzym Phospholipase A2 (PLA2) ermöglicht das. PLA2 wird durch zahlreiche Faktoren reguliert, z.B. wird sie aktiviert durch Zytokine, Wachstumsfaktoren und Bradykinin, gehemmt durch Glucocorticoide - diese erhöhen die Expression eines Annexins (Annexine - auch Lipokortine genannt - sind intrazelluläre Proteine, die PLA2 hemmen) und die Reduktion der Phospholipase A2-Aktivität wirkt entzündungshemmend.
Zyklooxygenasen (COX) produzieren Prostanoide (Prostaglandine und Thromboxane);
Lipoxygenasen (LOX) erzeugen Leukotriene und andere Metabolite; der Zytochrom-P450-Komplex oxydiert Arachidonsäure und daraus entstandene Metabolite. Die
Mischung der von einer Zelle produzierten Eikosanoide hängt von der
Aktivität ihrer Enzyme (Endoperoxid-Isomerasen, Synthasen) ab. So
entsteht in Thrombozyten vorwiegend TXA2, in Gefäßendothel PGI2.
Auch das Angebot an Substraten (z.B. Linolensäure, Arachidonsäure) -
und damit indirekt die Ernährung - beeinflusst das Muster der von den
Zellen synthetisierten Eikosanoide.
Eikosanoide
werden im Rahmen eines mehrstufigen Vorgangs rasch inaktiviert - zuerst
durch spezifische
Enzyme, welche die biologische Wirksamkeit beenden (Prostanoide:
Prostaglandin-Dehydrogenase - vor allem in der Lunge, Halbwertszeit der
meisten Prostaglandine im Kreislauf weniger als eine Minute, sowie
Reduktase), anschließend
durch Fettsäureoxidation. Die inaktiven Abbauprodukte werden mit dem
Harn ausgeschieden.
aktiviert durch Zytokine, Wachstumsfaktoren und Bradykinin, gehemmt durch Glucocorticoide - diese erhöhen die Expression eines Annexins (Annexine - auch Lipokortine genannt - sind intrazelluläre Proteine, die PLA2 hemmen) und die Reduktion der Phospholipase A2-Aktivität wirkt entzündungshemmend.
Zyklooxygenasen (COX) produzieren Prostanoide (Prostaglandine und Thromboxane);
Lipoxygenasen (LOX) erzeugen Leukotriene und andere Metabolite; der Zytochrom-P450-Komplex oxydiert Arachidonsäure und daraus entstandene Metabolite. Die
Mischung der von einer Zelle produzierten Eikosanoide hängt von der
Aktivität ihrer Enzyme (Endoperoxid-Isomerasen, Synthasen) ab. So
entsteht in Thrombozyten vorwiegend TXA2, in Gefäßendothel PGI2.
Auch das Angebot an Substraten (z.B. Linolensäure, Arachidonsäure) -
und damit indirekt die Ernährung - beeinflusst das Muster der von den
Zellen synthetisierten Eikosanoide.
Eikosanoide
werden im Rahmen eines mehrstufigen Vorgangs rasch inaktiviert - zuerst
durch spezifische
Enzyme, welche die biologische Wirksamkeit beenden (Prostanoide:
Prostaglandin-Dehydrogenase - vor allem in der Lunge, Halbwertszeit der
meisten Prostaglandine im Kreislauf weniger als eine Minute, sowie
Reduktase), anschließend
durch Fettsäureoxidation. Die inaktiven Abbauprodukte werden mit dem
Harn ausgeschieden.
 Prostanoide (Prostaglandine, Thromboxane), Leukotriene, Hydroxyeicosatetraensäuren (HETE; sie wirken auf neutrophile und eosinophile Granulozyten). Zu
Endocannabinoiden s. dort
Prostanoide (Prostaglandine, Thromboxane), Leukotriene, Hydroxyeicosatetraensäuren (HETE; sie wirken auf neutrophile und eosinophile Granulozyten). Zu
Endocannabinoiden s. dort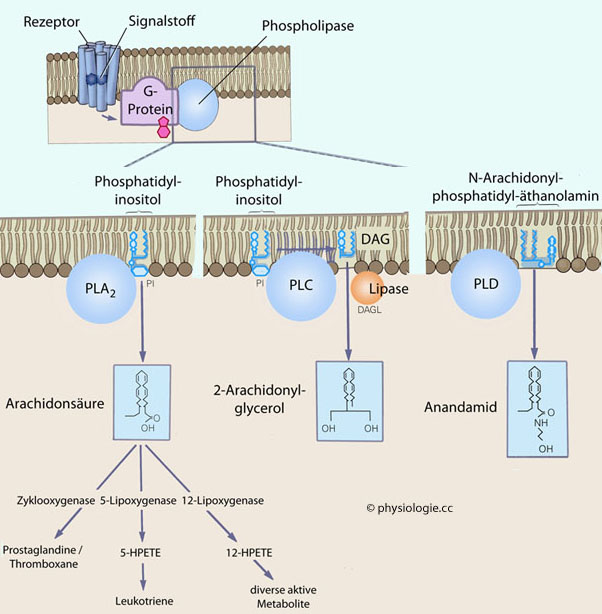 Abbildung: Durch Hydrolyse von Fettsäuren aus der Zellmembran bilden Phospholipasen spezifische second messengers PGD2 wirkt über DP1-Rezeptoren (Gs, cAMP-Erhöhung; Gefäßwände, Thrombozyten, ZNS, Luftwege, Auge) und DP2-Rezeptoren (cAMP-Senkung; weit verbreitet, insbesondere im Immunsystem). PGD2 wirkt in zahlreichen Gebieten vasodilatierend, relaxiert auch den Darm und den Uterus, verengt die Bronchien über eine sekundäre Wirkung auf TP-Rezeptoren und modifiziert die Freisetzung hypothalamisch-hypophysärer Hormone. PGE2 bewirkt PGF2α wirkt über FP-Rezeptoren (Gq/G11, PLC, Steigerung [Ca++]) wehenfördernd (antagonostisch zu PGE2). PGI2 (=Prostazyklin) wirkt über IP-Rezeptoren (Gs, cAMP-Erhöhung) aggregationshemmend auf Thrombozyten sowie vasodilatierend (es wirkt protektiv auf die Gefäßwand). TXA2 stimuliert TP-Rezeptoren (Gq/G11, PLC, Steigerung [Ca++]).
Es fördert die Plättchenaggregation und wirkt vaso- und
bronchokonstringierend. Hemmung der Thromboxansynthese hat eine
verlängerte Blutungszeit zur Folge.
Abbildung: Durch Hydrolyse von Fettsäuren aus der Zellmembran bilden Phospholipasen spezifische second messengers PGD2 wirkt über DP1-Rezeptoren (Gs, cAMP-Erhöhung; Gefäßwände, Thrombozyten, ZNS, Luftwege, Auge) und DP2-Rezeptoren (cAMP-Senkung; weit verbreitet, insbesondere im Immunsystem). PGD2 wirkt in zahlreichen Gebieten vasodilatierend, relaxiert auch den Darm und den Uterus, verengt die Bronchien über eine sekundäre Wirkung auf TP-Rezeptoren und modifiziert die Freisetzung hypothalamisch-hypophysärer Hormone. PGE2 bewirkt PGF2α wirkt über FP-Rezeptoren (Gq/G11, PLC, Steigerung [Ca++]) wehenfördernd (antagonostisch zu PGE2). PGI2 (=Prostazyklin) wirkt über IP-Rezeptoren (Gs, cAMP-Erhöhung) aggregationshemmend auf Thrombozyten sowie vasodilatierend (es wirkt protektiv auf die Gefäßwand). TXA2 stimuliert TP-Rezeptoren (Gq/G11, PLC, Steigerung [Ca++]).
Es fördert die Plättchenaggregation und wirkt vaso- und
bronchokonstringierend. Hemmung der Thromboxansynthese hat eine
verlängerte Blutungszeit zur Folge.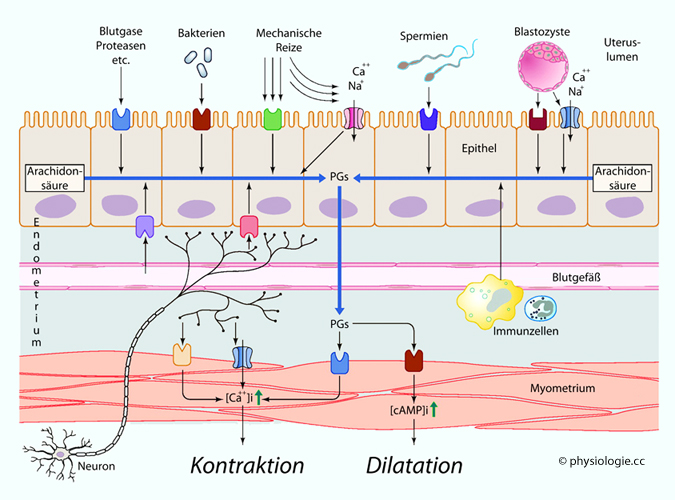 Abbildung: Prostaglandine beeinflussen den glattmuskulären Tonus Gefäßwand:
Das Effektprofil kann ganz unterschiedlich ausfallen, je nachdem, ob
die Gefäße physiologische Bedingungen vorfinden (COX-I: hämostatische
Wirkungen) oder ob eine Entzündungsreaktion vorliegt (COX-II:
Vasodilatation und Rötung, Erwärmung, Schwellung; sowie Schmerz- und
Fieberentstehung). TXA2, PGG2 und PGH2 bewirken TP-rezeptorvermittelte
Vasokonstriktion; diese kann aber durch vasodilatative Effekte von
PGI2, PGD und PGE überlagert sein. Diese erweitern über IP-, DP- und
EP-Rezeptoren die meisten Gefäßgebiete und wirken dadurch
blutdrucksenkend. Thrombozyten: PGI2 hemmt die Blutplättchen-aggregation. Im Gastrointestinaltrakt überwiegt ein Schutzeffekt
durch Anregung der Muzinsekretion und Dämpfung der gastralen
HCl- und Pepsinproduktion (PGE2, PGI2). Die vasodilatatorische Durchblutungsförderung wirkt in
dieselbe Richtung (PGE2, PGF2α). PGE und PGF kontrahieren (in vitro) die Längsmuskulatur, PGE2 relaxiert die Ringmuskulatur. Uterus: PGE2 und PGF2α werden wegen ihrer wehenanregenden Wirkung (Kraft und Frequenz) in der Geburtshilfe eingesetzt. Am nichtschwangeren Uterus wirkt PGE relaxierend, PGF anregend - die Effekte sind zyklusabhängig Nieren: Prostaglandine wirken vasodilatierend und stimulieren die Reninbildung. Die Salz-Wasser-Ausscheidung wird angeregt (PGE2, PGI2) Bronchien, Trachea: PGI2 und PGE1 relaxieren die glatte Muskulatur, TXA2 wirkt bronchokonstriktorisch
Abbildung: Prostaglandine beeinflussen den glattmuskulären Tonus Gefäßwand:
Das Effektprofil kann ganz unterschiedlich ausfallen, je nachdem, ob
die Gefäße physiologische Bedingungen vorfinden (COX-I: hämostatische
Wirkungen) oder ob eine Entzündungsreaktion vorliegt (COX-II:
Vasodilatation und Rötung, Erwärmung, Schwellung; sowie Schmerz- und
Fieberentstehung). TXA2, PGG2 und PGH2 bewirken TP-rezeptorvermittelte
Vasokonstriktion; diese kann aber durch vasodilatative Effekte von
PGI2, PGD und PGE überlagert sein. Diese erweitern über IP-, DP- und
EP-Rezeptoren die meisten Gefäßgebiete und wirken dadurch
blutdrucksenkend. Thrombozyten: PGI2 hemmt die Blutplättchen-aggregation. Im Gastrointestinaltrakt überwiegt ein Schutzeffekt
durch Anregung der Muzinsekretion und Dämpfung der gastralen
HCl- und Pepsinproduktion (PGE2, PGI2). Die vasodilatatorische Durchblutungsförderung wirkt in
dieselbe Richtung (PGE2, PGF2α). PGE und PGF kontrahieren (in vitro) die Längsmuskulatur, PGE2 relaxiert die Ringmuskulatur. Uterus: PGE2 und PGF2α werden wegen ihrer wehenanregenden Wirkung (Kraft und Frequenz) in der Geburtshilfe eingesetzt. Am nichtschwangeren Uterus wirkt PGE relaxierend, PGF anregend - die Effekte sind zyklusabhängig Nieren: Prostaglandine wirken vasodilatierend und stimulieren die Reninbildung. Die Salz-Wasser-Ausscheidung wird angeregt (PGE2, PGI2) Bronchien, Trachea: PGI2 und PGE1 relaxieren die glatte Muskulatur, TXA2 wirkt bronchokonstriktorisch Abbildung: Prostanoide beeinflussen den glattmuskulären Tonus COX1 ist konstitutiv und wird von den meisten Geweben exprimiert. Es wirkt homöostatisch in verschiedenen Geweben (Prostaglandine für die Zytoprotektion im Magen, für die Plättchenaggregation, Autoregulation in der Niere, Wehentätigkeit des Uterus).Abbildung): Weitere Schritte werden durch Synthasen bewerkstelligt, die nach ihrem Endprodukt benannt sind: PGD-Synthase, PGE-Synthase, PGF-Synthase, PGI-Synthase, TXA-Synthase. Die Produkte sind PGD2, PGE2, PGF2α, PGI2 und TXA2.
Abbildung: Prostanoide beeinflussen den glattmuskulären Tonus COX1 ist konstitutiv und wird von den meisten Geweben exprimiert. Es wirkt homöostatisch in verschiedenen Geweben (Prostaglandine für die Zytoprotektion im Magen, für die Plättchenaggregation, Autoregulation in der Niere, Wehentätigkeit des Uterus).Abbildung): Weitere Schritte werden durch Synthasen bewerkstelligt, die nach ihrem Endprodukt benannt sind: PGD-Synthase, PGE-Synthase, PGF-Synthase, PGI-Synthase, TXA-Synthase. Die Produkte sind PGD2, PGE2, PGF2α, PGI2 und TXA2. Störungen im sympathischen Nervensystem werden medikamentös durch Agonisten oder Antagonisten behandelt, d.h. Pharmaka, welche an den α- bzw. ß-Rezeptoren stimulierend bzw. inhibierend wirken. So verwendet man α-Adrenozeptor-Agonisten zur Vasokonstriktion und Blutdrucksteigerung, ß-Adrenozeptor-Agonisten zur Anregung der Herztätigkeit (ß1) oder zur Asthmatherapie (ß2). ß2-Blocker hingegen kommen
z.B. bei Herzinsuffizienz (Myokardschonung), koronarer Herzkrankheit
(Verminderung des Sauerstoffbedarfs) und Rhythmusstörungen zum Einsatz
(negativ dromotrope Wirkung). Schizophrenie (schizophrene Psychose), depressive Verstimmungen,
Aufmerksamkeitsdefizit- Hyperaktivitätssyndrom (Attention deficit hyperactivity disorder ADHS: Psychische Störung mit beeinträchtigter Aufmerksamkeit und Impulsivität sowie körperlicher Unruhe) oder Mb. Parkinson.
Störungen im sympathischen Nervensystem werden medikamentös durch Agonisten oder Antagonisten behandelt, d.h. Pharmaka, welche an den α- bzw. ß-Rezeptoren stimulierend bzw. inhibierend wirken. So verwendet man α-Adrenozeptor-Agonisten zur Vasokonstriktion und Blutdrucksteigerung, ß-Adrenozeptor-Agonisten zur Anregung der Herztätigkeit (ß1) oder zur Asthmatherapie (ß2). ß2-Blocker hingegen kommen
z.B. bei Herzinsuffizienz (Myokardschonung), koronarer Herzkrankheit
(Verminderung des Sauerstoffbedarfs) und Rhythmusstörungen zum Einsatz
(negativ dromotrope Wirkung). Schizophrenie (schizophrene Psychose), depressive Verstimmungen,
Aufmerksamkeitsdefizit- Hyperaktivitätssyndrom (Attention deficit hyperactivity disorder ADHS: Psychische Störung mit beeinträchtigter Aufmerksamkeit und Impulsivität sowie körperlicher Unruhe) oder Mb. Parkinson.

 Acetylcholin
wirkt als Transmitter für alle aus dem ZNS austretenden Neurone.
Cholinerg sind motorische Endplatten, präganglionäre Fasern,
postganglionär parasympathische sowie sudomotorische sympathische
Nervernfasern, einige Neurone im ZNS. Nikotinische und muskarinische Rezeptoren können sowohl exzitatorisch als auch inhibitorisch wirken. Nikotinische Rezeptoren sind ligandengesteuerte Ionenkanäle (Na+, Ca++, K+), sie bewirken Endplattenpotentiale (Muskeltyp) und EPSPs (neuronaler Typ). Muskarinische sind G-Protein-gekoppelte Rezeptoren,
die Gq (M1, M3, M5) oder Gi aktivieren (M2, M4). M1 ("neural") fördern
die Erregung (Gehirn); M2 ("kardial") wirken auf Herz,
Harnblase, Ureter; M3 ("glattmuskulär") auf Auge, gastrointestinales System,
Bronchien; alle Typen finden sich im Gehirn. Freigesetztes Acetylcholin
bindet an postsynaptische und präsynaptische Rezeptoren oder wird durch
membranständige Acetylcholinesterase abgebaut Purine wirken über ionotrope (P2X) und metabotrope Rezeptoren (P2Y, P2U) z.B. auf Herz, Blutplättchen, Immun- und Nervensystem. ATP wird von postganglionär-sympathischen Fasern zusammen mit Noradrenalin aus synaptischen Vesikeln ausgeschüttet (Kotransmission) und wirkt vasokonstriktorisch, wobei Ca++-Einstrom in mehreren Phasen erfolgt. Die
Intensität der Erregungsstärke an sympathischen Varikositäten wird über ATP in eine verstärkte Kontraktion der
Muskelzelle übersetzt. ATP ist auch Kotransmitter in
cholinergen Nerven und im Darmnervensystem und wirkt schmerz- und
entzündungsauslösend. Adenosin wirkt über Adenosinrezeptoren auf Herzmuskel (anti-dysrhythmisch),
Gefäße (vasodilatierend), Gehirn (sedierend, krampflösend,
schmerzstillend, protektiv), Fettgewebe (lipolytisch), Leber
(glukoneogenetisch) Katecholamine (Dopamin, Noradrenalin, Adrenalin) werden über DOPA aus Tyrosin gebildet - Tyrosinhydroxylase (Tyrosin → DOPA) ist das geschwindigkeitslimitierende Enzym -, in Varikositäten gespeichert, nach ihrer Freisetzung Na+-abhängig wiederverwertet (Neurit) oder abgebaut (Glia). Der Abbau erfolgt mittels Monoaminooxidase (MAO, äußere Mitochondrienmembran) sowie Catechol-O- Methlytransferase (COMT, zytoplasmatisch), die Halbwertszeit ist kurz. Die wichtigsten Abbauprodukte sind Vanillinmandelsäure und
Homovanillinsäure, ihre Ausscheidung (Harn) ist ein Maß für die Katecholaminproduktion im Körper. Dopamin wirkt über Dopaminrezeptoren: Na+-, Ca++-Kanäle (D1-Gruppe), K+-Kanäle
(D2-Gruppe), ligandengesteuerte Kanäle (glutamaterge Rezeptoren NMDA,
AMPA). Dopaminerge Neurone sind in Bewegungskontrolle (75%: Striatum),
Gewöhnung, Belohnung, Motivation, Erinnerung, Vergessen, Übelkeit
involviert und der wichtigste Inhibitor der Prolaktinsekretion. Dopamin
steigert die Natriurese, die Wirkung auf Blutgefäße ist dosisabhängig. - Postsynaptische sympathische Fasern sezernieren zu ~95% Noradrenalin (Wirkung auf α- und ß1-Rezeptoren), die Nebenniere zu ~80% Adrenalin (an ß-Rezeptoren wirksamer als Noradrenalin). α1-Rezeptoren
vermitteln alle sympathischen Effekte, die auf Kontraktion glatter
Muskulatur beruhen, α2-Rezeptoren u.a. die präsynaptische Selbsthemmung
der Noradrenalinfreisetzung. α1
aktivieren Phospholipase C (→ IP3, DAG), α2 hemmen die Adenylylcyclase
(cAMP↓), β-Rezeptoren regen sie an (cAMP↑). ß1-Rezeptoren wirken
positiv auf die Herzqualitäten, ß2-Rezeptoren entspannen Luftwege,
Koronararterien, Skelettmuskelarteriolen, Darm, Blasenmuskel, Uterus,
Samenleiter, Ziliarmuskel; ß3-Rezeptoren vermitteln Lipolyse
(Fettgewebe) und Thermogenese (Skelettmuskel) Biogene Amine entstehen durch Decarboxylierung von Aminosäuren - Histamin aus Histidin, Serotonin aus 5-Hydroxytryptophan. Histamin wirkt
über G-Protein-mediierte Rezeptoren (H1 bis H4): H1 aktivieren
Phospholipase C, H2 Adenylylcyclase, H3 und H4 hemmen Adenylylcyclase.
Histamin wirkt im ZNS (Schlaf-Wach-Regulation, Lernen und Gedächtnis,
Immunität, Nahrungs- und Wasseraufnahme, Temperaturregulation), Magen
(Salzsäureproduktion, Pepsinogenfreisetzung, Peristaltik), Bronchien
(Konstriktion), Gefäßen (Dilatation, Permeabilität), Gewebe (Mastzellendegranulation: Schmerz, Entzündung). - Serotonin findet sich zu 90% in ECL (enterochromaffinähnlichen) Zellen des Darms. Es wirkt
über 5-HT-Rezeptoren: Der 5-HT3-Rezeptor ist ionotrop, die anderen sind
metabotrop. 5-HT1-Rezeptoren hemmen die Adenylylcyclase, senken den
Blutdruck, wirken anxiolytisch, kontrahieren Blutgefäße in Meningen und
Koronarien, wirken im Trigeminusgebiet schmerzhemmend; 5-HT2-Rezeptoren
kontrahieren die Magen-Darm-Muskulatur, aktivieren Thrombozyten, haben
psychotrope Wirkung, setzen NO aus Endothelien frei, wirken
appetitzügelnd; 5-HT3-Rezeptoren (M-Rezeptoren) sind Ionenkanäle und
sind an Reflexen beteiligt; 5-HT4-Rezeptoren wirken
peristaltikfördernd, positiv inotrop und chronotrop. Das Gehirn exprimiert alle Arten von Serotoninrezeptoren; hier
wirkt Serotonin als Neuromodulator und steuert Schlafmuster.
Serotonin-Wiederaufnahme-Hemmer (SSRI) erhöhen die Serotoninmenge im
Synapsenspalt Zur Gruppe der Peptide von
Signalstoffen gehören die Mehrzahl der Hormone, Tachykinine und
Endotheline. Zur Gruppe der Tachykinine gehören Neurotransmitter und
Gewebshormone (Substanz P), die glatte Muskulatur über Tachykininrezeptoren (NK1, NK2 und NK3) rasch kontrahieren lassen. - Endotheline (ET1, ET2, ET3) wirken über zwei Arten von Rezeptoren: ETA bewirken Natriumretention und Blutdruckerhöhung (glatte Gefäßmuskelzellen), ETB Natriurese und Blutdrucksenkung (Endothelzellen: Freisetzung von Prostazyklin und NO). Viele Zellarten verfügen über ET-Rezeptoren (Gefäße, Herz, Nebenniere, Sympathikus),
das Wirkungsprofil ist komplex. ET-1 spielt unter
physiologischen Bedingungen eine eher geringe Rolle, bei geschädigtem
Endothel treten seine vasokonstriktorischen Effekte hervor. - Der
Kotransmitter VIP (vasoaktives intestinales Peptid)
aus cholinergen Endigungen, neuroendokrinen Zellen und pankreatischen
D-Zellen hyperpolarisiert glatte Muskelzellen der
Gefäßwand, wirkt auf zirkadiane Rhythmen, hemmt Darmmotorik, vermehrt
Bicarbonatsekretion und wirkt positiv inotrop, vasodilatierend,
blutdrucksenkend. - NPY (Neuropeptid Y) wirkt neurogenetisch (Gehirn) und vasokonstriktorisch. - Substanz P
aus Neuronen und Leukozyten unterstützt über NK1-Rezeptoren
Vasodilatation und Filtration, Chemotaxis von Leukozyten,
Schmerzentstehung und Wundheilung; über NK2-Rezeptoren Darmmotorik;
über NK3-Rezeptoren die Freisetzung von Acetylcholin an cholinergen
Nervenfasern Gasotransmitter
werden in Zellen hergestellt, diffundieren leicht und wirken in
niedriger Dosis lokal als Signalmoleküle. NO
(Stickstoffmonoxid) wird durch NO-Synthasen (NOS) aus L-Arginin
freigesetzt: Endotheliale (eNOS oder NOS3, aktivierbar durch
Scherkräfte, bewirkt Vasodilatation / Blutdrucksenkung), neuronale (nNOS oder NOS, in ~2% aller Neurone im Gehirn nachweisbar, verstärkt die glutamaterge Neurotransmission)
und induzierbare (iNOS oder NOS2). NO wird innerhalb von
Sekunden abgebaut. - CO
(Kohlenmonoxid) entsteht beim Abbau von Hämoglobin durch
Hämoxygenase-1 (Leber, Milz) und Hämoxygenase-2 (Gehirn, Endothelien). CO relaxiert glatte Gefäßmuskulatur (Perfusion +, Blutdruck -), moduliert zirkadiane Rhythmen und ist an der Geruchswahrnehmung beteiligt. - H2S
(Schwefelwasserstoff) wird enzymatisch aus Zystein freigesetzt, wirkt
vasodilatierend und perfusionssteigernd, über den Hypothalamus
blutdrucksenkend und dämpfend auf die Stressachse; es erhöht die
Glutathionsynthese und inaktiviert Sauerstoffsradikale
(Oxidationsschutz) Phospholipase A2 setzt auf physikalische, chemische oder hormonelle (Zytokine, Wachstumsfaktoren) Reize hin Arachidonsäure aus der Zellmembran frei, spezifische Enzyme bilden daraus Eikosanoide (Prostaglandine, Prostazyklin, Thromboxan, Leukotriene etc). Cyklooxygenase (COX) bildet Prostanoide (Prostaglandine und Thromboxane), Lipoxygenase (LOX) Leukotriene. PGD-Synthase bildet PDG2, PGE-Synthase PGE2 etc. Eikosanoide
werden rasch inaktiviert - durch spezifische Enzyme und durch
Oxidation. PGD2 verengt Bronchien und wirkt schlaffördernd, PGE2
dilatiert Gefäße und Bronchien, fördert die Schleimbildung im Gastrointestinaltrakt und entspannt den Uterus, PGF2α
kontrahiert Bronchien, Gefäße und Uterus, PGI2
(=Prostazyklin) wirkt vasodilatierend und aggregationshemmend auf
Thrombozyten, TXA2 stimuliert die Plättchenaggregation, wirkt vaso- und
bronchokonstingierend. Insgesamt ist das Effektprofil an Gefäßen je
nach Situation unterschiedlich; im Gastrointestinaltrakt überwiegt ein
Schutzeffekt durch Anregung der Muzinsekretion und Dämpfung der HCl-
und Pepsinproduktion; im Uterus sind die Effekte zyklusabhängig; in der
Niere wirken Prostaglandine vasodilatierend, stimulieren die
Reninbildung und regen die Salz-Wasser-Ausscheidung an; die Bronchien
werden durch PGI2 und PGE1 relaxiert, durch TXA2 konstringiert Acetylcholin
wirkt als Transmitter für alle aus dem ZNS austretenden Neurone.
Cholinerg sind motorische Endplatten, präganglionäre Fasern,
postganglionär parasympathische sowie sudomotorische sympathische
Nervernfasern, einige Neurone im ZNS. Nikotinische und muskarinische Rezeptoren können sowohl exzitatorisch als auch inhibitorisch wirken. Nikotinische Rezeptoren sind ligandengesteuerte Ionenkanäle (Na+, Ca++, K+), sie bewirken Endplattenpotentiale (Muskeltyp) und EPSPs (neuronaler Typ). Muskarinische sind G-Protein-gekoppelte Rezeptoren,
die Gq (M1, M3, M5) oder Gi aktivieren (M2, M4). M1 ("neural") fördern
die Erregung (Gehirn); M2 ("kardial") wirken auf Herz,
Harnblase, Ureter; M3 ("glattmuskulär") auf Auge, gastrointestinales System,
Bronchien; alle Typen finden sich im Gehirn. Freigesetztes Acetylcholin
bindet an postsynaptische und präsynaptische Rezeptoren oder wird durch
membranständige Acetylcholinesterase abgebaut Purine wirken über ionotrope (P2X) und metabotrope Rezeptoren (P2Y, P2U) z.B. auf Herz, Blutplättchen, Immun- und Nervensystem. ATP wird von postganglionär-sympathischen Fasern zusammen mit Noradrenalin aus synaptischen Vesikeln ausgeschüttet (Kotransmission) und wirkt vasokonstriktorisch, wobei Ca++-Einstrom in mehreren Phasen erfolgt. Die
Intensität der Erregungsstärke an sympathischen Varikositäten wird über ATP in eine verstärkte Kontraktion der
Muskelzelle übersetzt. ATP ist auch Kotransmitter in
cholinergen Nerven und im Darmnervensystem und wirkt schmerz- und
entzündungsauslösend. Adenosin wirkt über Adenosinrezeptoren auf Herzmuskel (anti-dysrhythmisch),
Gefäße (vasodilatierend), Gehirn (sedierend, krampflösend,
schmerzstillend, protektiv), Fettgewebe (lipolytisch), Leber
(glukoneogenetisch) Katecholamine (Dopamin, Noradrenalin, Adrenalin) werden über DOPA aus Tyrosin gebildet - Tyrosinhydroxylase (Tyrosin → DOPA) ist das geschwindigkeitslimitierende Enzym -, in Varikositäten gespeichert, nach ihrer Freisetzung Na+-abhängig wiederverwertet (Neurit) oder abgebaut (Glia). Der Abbau erfolgt mittels Monoaminooxidase (MAO, äußere Mitochondrienmembran) sowie Catechol-O- Methlytransferase (COMT, zytoplasmatisch), die Halbwertszeit ist kurz. Die wichtigsten Abbauprodukte sind Vanillinmandelsäure und
Homovanillinsäure, ihre Ausscheidung (Harn) ist ein Maß für die Katecholaminproduktion im Körper. Dopamin wirkt über Dopaminrezeptoren: Na+-, Ca++-Kanäle (D1-Gruppe), K+-Kanäle
(D2-Gruppe), ligandengesteuerte Kanäle (glutamaterge Rezeptoren NMDA,
AMPA). Dopaminerge Neurone sind in Bewegungskontrolle (75%: Striatum),
Gewöhnung, Belohnung, Motivation, Erinnerung, Vergessen, Übelkeit
involviert und der wichtigste Inhibitor der Prolaktinsekretion. Dopamin
steigert die Natriurese, die Wirkung auf Blutgefäße ist dosisabhängig. - Postsynaptische sympathische Fasern sezernieren zu ~95% Noradrenalin (Wirkung auf α- und ß1-Rezeptoren), die Nebenniere zu ~80% Adrenalin (an ß-Rezeptoren wirksamer als Noradrenalin). α1-Rezeptoren
vermitteln alle sympathischen Effekte, die auf Kontraktion glatter
Muskulatur beruhen, α2-Rezeptoren u.a. die präsynaptische Selbsthemmung
der Noradrenalinfreisetzung. α1
aktivieren Phospholipase C (→ IP3, DAG), α2 hemmen die Adenylylcyclase
(cAMP↓), β-Rezeptoren regen sie an (cAMP↑). ß1-Rezeptoren wirken
positiv auf die Herzqualitäten, ß2-Rezeptoren entspannen Luftwege,
Koronararterien, Skelettmuskelarteriolen, Darm, Blasenmuskel, Uterus,
Samenleiter, Ziliarmuskel; ß3-Rezeptoren vermitteln Lipolyse
(Fettgewebe) und Thermogenese (Skelettmuskel) Biogene Amine entstehen durch Decarboxylierung von Aminosäuren - Histamin aus Histidin, Serotonin aus 5-Hydroxytryptophan. Histamin wirkt
über G-Protein-mediierte Rezeptoren (H1 bis H4): H1 aktivieren
Phospholipase C, H2 Adenylylcyclase, H3 und H4 hemmen Adenylylcyclase.
Histamin wirkt im ZNS (Schlaf-Wach-Regulation, Lernen und Gedächtnis,
Immunität, Nahrungs- und Wasseraufnahme, Temperaturregulation), Magen
(Salzsäureproduktion, Pepsinogenfreisetzung, Peristaltik), Bronchien
(Konstriktion), Gefäßen (Dilatation, Permeabilität), Gewebe (Mastzellendegranulation: Schmerz, Entzündung). - Serotonin findet sich zu 90% in ECL (enterochromaffinähnlichen) Zellen des Darms. Es wirkt
über 5-HT-Rezeptoren: Der 5-HT3-Rezeptor ist ionotrop, die anderen sind
metabotrop. 5-HT1-Rezeptoren hemmen die Adenylylcyclase, senken den
Blutdruck, wirken anxiolytisch, kontrahieren Blutgefäße in Meningen und
Koronarien, wirken im Trigeminusgebiet schmerzhemmend; 5-HT2-Rezeptoren
kontrahieren die Magen-Darm-Muskulatur, aktivieren Thrombozyten, haben
psychotrope Wirkung, setzen NO aus Endothelien frei, wirken
appetitzügelnd; 5-HT3-Rezeptoren (M-Rezeptoren) sind Ionenkanäle und
sind an Reflexen beteiligt; 5-HT4-Rezeptoren wirken
peristaltikfördernd, positiv inotrop und chronotrop. Das Gehirn exprimiert alle Arten von Serotoninrezeptoren; hier
wirkt Serotonin als Neuromodulator und steuert Schlafmuster.
Serotonin-Wiederaufnahme-Hemmer (SSRI) erhöhen die Serotoninmenge im
Synapsenspalt Zur Gruppe der Peptide von
Signalstoffen gehören die Mehrzahl der Hormone, Tachykinine und
Endotheline. Zur Gruppe der Tachykinine gehören Neurotransmitter und
Gewebshormone (Substanz P), die glatte Muskulatur über Tachykininrezeptoren (NK1, NK2 und NK3) rasch kontrahieren lassen. - Endotheline (ET1, ET2, ET3) wirken über zwei Arten von Rezeptoren: ETA bewirken Natriumretention und Blutdruckerhöhung (glatte Gefäßmuskelzellen), ETB Natriurese und Blutdrucksenkung (Endothelzellen: Freisetzung von Prostazyklin und NO). Viele Zellarten verfügen über ET-Rezeptoren (Gefäße, Herz, Nebenniere, Sympathikus),
das Wirkungsprofil ist komplex. ET-1 spielt unter
physiologischen Bedingungen eine eher geringe Rolle, bei geschädigtem
Endothel treten seine vasokonstriktorischen Effekte hervor. - Der
Kotransmitter VIP (vasoaktives intestinales Peptid)
aus cholinergen Endigungen, neuroendokrinen Zellen und pankreatischen
D-Zellen hyperpolarisiert glatte Muskelzellen der
Gefäßwand, wirkt auf zirkadiane Rhythmen, hemmt Darmmotorik, vermehrt
Bicarbonatsekretion und wirkt positiv inotrop, vasodilatierend,
blutdrucksenkend. - NPY (Neuropeptid Y) wirkt neurogenetisch (Gehirn) und vasokonstriktorisch. - Substanz P
aus Neuronen und Leukozyten unterstützt über NK1-Rezeptoren
Vasodilatation und Filtration, Chemotaxis von Leukozyten,
Schmerzentstehung und Wundheilung; über NK2-Rezeptoren Darmmotorik;
über NK3-Rezeptoren die Freisetzung von Acetylcholin an cholinergen
Nervenfasern Gasotransmitter
werden in Zellen hergestellt, diffundieren leicht und wirken in
niedriger Dosis lokal als Signalmoleküle. NO
(Stickstoffmonoxid) wird durch NO-Synthasen (NOS) aus L-Arginin
freigesetzt: Endotheliale (eNOS oder NOS3, aktivierbar durch
Scherkräfte, bewirkt Vasodilatation / Blutdrucksenkung), neuronale (nNOS oder NOS, in ~2% aller Neurone im Gehirn nachweisbar, verstärkt die glutamaterge Neurotransmission)
und induzierbare (iNOS oder NOS2). NO wird innerhalb von
Sekunden abgebaut. - CO
(Kohlenmonoxid) entsteht beim Abbau von Hämoglobin durch
Hämoxygenase-1 (Leber, Milz) und Hämoxygenase-2 (Gehirn, Endothelien). CO relaxiert glatte Gefäßmuskulatur (Perfusion +, Blutdruck -), moduliert zirkadiane Rhythmen und ist an der Geruchswahrnehmung beteiligt. - H2S
(Schwefelwasserstoff) wird enzymatisch aus Zystein freigesetzt, wirkt
vasodilatierend und perfusionssteigernd, über den Hypothalamus
blutdrucksenkend und dämpfend auf die Stressachse; es erhöht die
Glutathionsynthese und inaktiviert Sauerstoffsradikale
(Oxidationsschutz) Phospholipase A2 setzt auf physikalische, chemische oder hormonelle (Zytokine, Wachstumsfaktoren) Reize hin Arachidonsäure aus der Zellmembran frei, spezifische Enzyme bilden daraus Eikosanoide (Prostaglandine, Prostazyklin, Thromboxan, Leukotriene etc). Cyklooxygenase (COX) bildet Prostanoide (Prostaglandine und Thromboxane), Lipoxygenase (LOX) Leukotriene. PGD-Synthase bildet PDG2, PGE-Synthase PGE2 etc. Eikosanoide
werden rasch inaktiviert - durch spezifische Enzyme und durch
Oxidation. PGD2 verengt Bronchien und wirkt schlaffördernd, PGE2
dilatiert Gefäße und Bronchien, fördert die Schleimbildung im Gastrointestinaltrakt und entspannt den Uterus, PGF2α
kontrahiert Bronchien, Gefäße und Uterus, PGI2
(=Prostazyklin) wirkt vasodilatierend und aggregationshemmend auf
Thrombozyten, TXA2 stimuliert die Plättchenaggregation, wirkt vaso- und
bronchokonstingierend. Insgesamt ist das Effektprofil an Gefäßen je
nach Situation unterschiedlich; im Gastrointestinaltrakt überwiegt ein
Schutzeffekt durch Anregung der Muzinsekretion und Dämpfung der HCl-
und Pepsinproduktion; im Uterus sind die Effekte zyklusabhängig; in der
Niere wirken Prostaglandine vasodilatierend, stimulieren die
Reninbildung und regen die Salz-Wasser-Ausscheidung an; die Bronchien
werden durch PGI2 und PGE1 relaxiert, durch TXA2 konstringiert |

