

Eine Reise durch die Physiologie - Wie der Körper des Menschen funktioniert

Synapsen
© H. Hinghofer-Szalkay
 Clathrin: clatri (lat) = Gitter, Käfig
Clathrin: clatri (lat) = Gitter, Käfig
Dendrit: δενδρίτης = verzweigt, von Bäumen abstammend (δένδρον = Baum)
De-, Hyperpolarisation: de = von..weg, ὑπέρ = über..hinaus, πολος = Achse(npunkt)
Gephyrin: γἑφυϱα = Brücke
Glia: γλία = Leim, Kitt
Glycin: γλυκύς = süß
SNARE: für soluble N-ethylmaleimide-sensitive fusion attachment protein receptor
Synapse: σύν = zusammen, ἅπτειν = fassen, ergreifen, συναψις = Verbindung
Triskele: τρισκελής = dreibeinig
Für die Kommunikation zwischen Nervenzellen verfügt ein erwachsener Mensch über etwa
hundert Billionen (~1014) Synapsen. Ihre
primäre Wirkung ist eine Veränderung des Zustands der nachgeschalteten (postsynaptischen, "empfangenden") Zelle im Sinne einer Verstärkung (inhibitorisches postsynaptisches Potential, IPSP) oder Abschwächung (exzitatorisches postsynaptisches Potential, EPSP) des Membranpotentials: IPSPs erschweren, EPSPs erleichtern die Entstehung eines Aktionspotentials am postsynaptischen Neuron.
Das präsynaptische
("sendende") Neuron synthetisiert Neurotransmitter in der Nähe des Zellkerns, transportiert den Transmitter durch den Neurit, speichert ihn in Vesikeln und
sezerniert ihn schließlich über Exozytose. Die Exozytose erfolgt mittels vesikulärer Proteinkomplexe des
SNARE-Mechanismus (soluble N-ethylmaleimide- sensitive- factor attachment receptor).
Transmitter
diffundieren über den synaptischen Spaltraum (~20 nm) und binden an
postsynaptische Rezeptoren. Dies löst rezeptortypische Folgereaktionen (z.B.
Natriumeinstrom und Depolarisation) aus, welche postsynaptische Auswirkungen haben.
Je nach Transmitter erfolgt rasche (Millisekundenbereich: Glutamat, GABA, Glycin, Acetylcholin nikotinerg..) oder langsame Übertragung (Sekundenbereich: Katecholamine, Acetylcholin muskarinerg).
Kotransmitter bewirken zusätzlich Fazilitation oder Depression (Sekunden- bis Minutenbereich oder länger) sowie Modulation des synaptischen Effekts (Sekunden bis Tage: Neuropeptide).
|
Synapsen & Transmitter  Neurotransmitter Vesikelzyklus, Speicherung, Freisetzung und Recycling, SNARE-Proteine Synaptischer Spaltraum Postsynaptischer Apparat präsynaptische vs. postsynaptische Rezeptoren Neuromodulation Postsynaptisaches Potential und Summation, EPSP und IPSP, Genexpression Entfernung des Transmitters aus dem synaptischen Spaltraum Glutamat GABA Glycin Bahnung und Hemmung Colokalisation und Cotransmission Konvergenz & Divergenz
Neurotransmitter Vesikelzyklus, Speicherung, Freisetzung und Recycling, SNARE-Proteine Synaptischer Spaltraum Postsynaptischer Apparat präsynaptische vs. postsynaptische Rezeptoren Neuromodulation Postsynaptisaches Potential und Summation, EPSP und IPSP, Genexpression Entfernung des Transmitters aus dem synaptischen Spaltraum Glutamat GABA Glycin Bahnung und Hemmung Colokalisation und Cotransmission Konvergenz & Divergenz

Neurotransmitter  Synapsine CaMKII Synaptotagmine Synaptobrevin Aktive Zone Rab3 Clathrin Fazilitation, Depression, Habituation, Augmentation Neuromodulation Disinhibition Cotransmission
Synapsine CaMKII Synaptotagmine Synaptobrevin Aktive Zone Rab3 Clathrin Fazilitation, Depression, Habituation, Augmentation Neuromodulation Disinhibition Cotransmission
 Praktische Aspekte
Praktische Aspekte  Core messages
Core messages

 Über Acetylcholin, Katecholamine, Serotonin, Histamin, ATP, Opioide und Prostaglandine s. dort
Über Acetylcholin, Katecholamine, Serotonin, Histamin, ATP, Opioide und Prostaglandine s. dort
Über zentralnervöse noradrenerge, serotoninerge, dopaminerge und cholinerge Systeme s. dort
Nervenzellen
verständigen sich mittels Neurotransmittern untereinander -
präsynaptische Zellen senden diese aus, postsynaptische reagieren
darauf. Neurotransmitter können kleine (Amine, Aminosäuren, Purine)
oder größere Moleküle (Peptide) sein. Über 90% aller Synapsen des
Gehirns nutzen Glutamat als Transmitterstoff.
Wie kommunizieren Nervenzellen?
Über die Arbeitsweise der Gehirns s. dort
Die Gesamtzahl der Nervenzellen
(Neuronen) im Körper eines Erwachsenen wird auf 86 Milliarden
geschätzt, davon mehr als die Hälfte in der Kleinhirnrinde und weniger
als 20% in der Großhirnrinde. Sie sind über Synapsen  miteinander verschaltet (Gesamtzahl im Körper ~1014),
wobei eine Nervenzelle auf bis zu ~5.104 andere synaptisch wirken
(Divergenz) und umgekehrt von bis zu ~5.104 anderen synaptisch erreicht werden
kann (Konvergenz).
Je komplexer die Verschaltungsmuster - sowohl zwischen einzelnen Zellen
als auch insgesamt im Verbund, also im Gehirn -, desto größer die
Wahrscheinlichkeit, dass die Aktivität solcher komplexen Systeme
(welche auch Sinnesmeldungen - aus der Umwelt und aus dem Körperinneren
- einbezieht) etwas ergibt, was man als Bewusstsein bezeichnet.
miteinander verschaltet (Gesamtzahl im Körper ~1014),
wobei eine Nervenzelle auf bis zu ~5.104 andere synaptisch wirken
(Divergenz) und umgekehrt von bis zu ~5.104 anderen synaptisch erreicht werden
kann (Konvergenz).
Je komplexer die Verschaltungsmuster - sowohl zwischen einzelnen Zellen
als auch insgesamt im Verbund, also im Gehirn -, desto größer die
Wahrscheinlichkeit, dass die Aktivität solcher komplexen Systeme
(welche auch Sinnesmeldungen - aus der Umwelt und aus dem Körperinneren
- einbezieht) etwas ergibt, was man als Bewusstsein bezeichnet.
Man unterscheidet zwei Arten von Synapsen: Chemische (bei weitem die Mehrzahl im
Gehirn des Menschen) und elektrische (über gap junctions). Die folgende Tabelle zeigt, worin
sie sich unterscheiden:
Vergleich elektrische - chemische Synapsen
Nach Kandel / Koester / Mack / Siegelbaum (eds), Principles of Neural Sciences, 6th ed. 2021 (McGraw Hill)
|
Typ
Synapse
|
Abstand prä- post- synaptisch
|
Zytoplas-
matische Kontinuität?
|
Ultra-
struktur
|
Trans-
mission durch
|
Synaptische Verzögerung
|
Richtung der Übertragung
|
Elektrisch
|
4 nm
|
ja
|
Gap junction- Kanäle
|
Ionenstrom
|
nein
|
meist bidirektional
|
Chemisch
|
20-40 nm
|
nein
|
Vesikel (präsyn.)
Rezeptoren
(postsyn.)
|
chemische Transmitter
|
≥0,3 ms
(meist 1-5 ms)
|
unidirektional
("Einbahn")
|
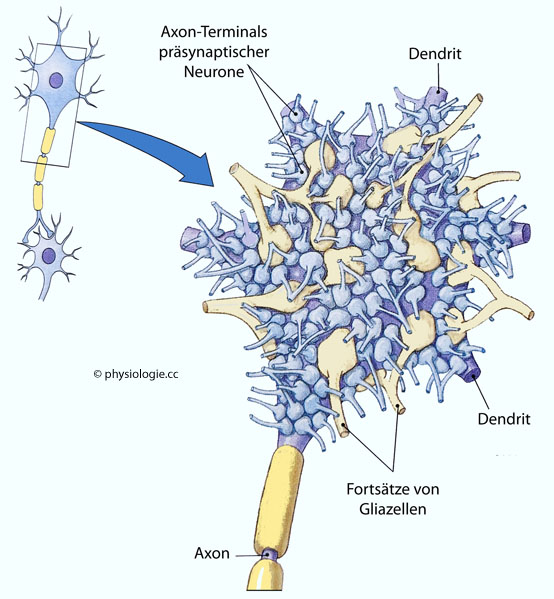

Abbildung: Soma eines postsynaptischen Neurons
Nach einer Vorlage bei Silverthorn, Human Physiology, an integrated approach, 4th Int'l ed. 2007, Pearson / Benjamin Cummings
Der
Zellkörper (das Soma) einer Nervenzelle ist fast vollständig von
Axonendigungen (Terminals) anderer Nervenzellen (präsynaptischer
Neurone) bedeckt. Dadurch kann dieses Neuron synaptische Impulse
hunderter anderer Neurone empfangen.
Dazu kommen zehlreiche Fortsätze von Gliazellen,
welche die Nervenzelle mechanisch stützen, zu ihrer elektrischen
Isolierung beitragen, Sauerstoff und Nährstoffe bereitstellen,
freigesetzten Transmitterstoff aufnehmen und recyceln, Pathogene
bekämpfen sowie Reste abgestorbener Nervenzellen entsorgen können
 Während sich bei elektrischen
Synapsen Änderungen des Membranpotentials (De- oder Hyperpolarisierung) der einen Zelle mittels gap junctions auf die Nachbarzelle (abgeschwächt) direkt und extrem rasch auswirken (elektrotonische Transmission) und deren Membranpotential beeinflussen (vorteihaft z.B. für Synchronisierungen), gibt es bei
chemischen Synapsen keine
solche elektrische Brücke. Vielmehr triggert ein präsynaptisches
Aktionspotential an der chemischen Synapse die Freisetzung eines Transmitterstoffs, der dann über Rezeptoren de- oder hyperpolarisierend wirkt - je nach
dem Effekt, den diese postsynaptisch auslösen.
Chemischen Synapsen stehen molekulare Verstärkungsmechanismen
zur Verfügung, die elektrische Synapsen nicht haben: Bildung /
Freisetzung von second messengers, Aktivierung von Kinasen - bei diesen
Schritten multipliziert sich der postsynaptische Effekt. Außerdem
können chemische Synapsen sowohl exzitatorisch als auch inhibitorisch
wirken, also die "Polung" des Informationsflusses ändern. Ein weiterer
Vorteil ist die Tatsache, dass sich die Aktivierung chemischer Synapsen
auf den Zustand von Nervenzelklen über längere Zeit (bis zu Stunden) auswirken kann.
Synapsen im Nervensystem unterscheiden sich - bei ähnlichem Funktionsprinzip - in einigen Eigenschaften von motorischen Endplatten.
Ähnlich gibt es auch innerhalb des ZNS Unterschiede zwischen speziellen
Synapsentypen. Dies trägt zur Diversität neurophysiologischer Spezifika
im Gehirn bei. Die prinzipiellen Schritte bei der Funktion einer
chemischen Synapse sind die folgenden:
Während sich bei elektrischen
Synapsen Änderungen des Membranpotentials (De- oder Hyperpolarisierung) der einen Zelle mittels gap junctions auf die Nachbarzelle (abgeschwächt) direkt und extrem rasch auswirken (elektrotonische Transmission) und deren Membranpotential beeinflussen (vorteihaft z.B. für Synchronisierungen), gibt es bei
chemischen Synapsen keine
solche elektrische Brücke. Vielmehr triggert ein präsynaptisches
Aktionspotential an der chemischen Synapse die Freisetzung eines Transmitterstoffs, der dann über Rezeptoren de- oder hyperpolarisierend wirkt - je nach
dem Effekt, den diese postsynaptisch auslösen.
Chemischen Synapsen stehen molekulare Verstärkungsmechanismen
zur Verfügung, die elektrische Synapsen nicht haben: Bildung /
Freisetzung von second messengers, Aktivierung von Kinasen - bei diesen
Schritten multipliziert sich der postsynaptische Effekt. Außerdem
können chemische Synapsen sowohl exzitatorisch als auch inhibitorisch
wirken, also die "Polung" des Informationsflusses ändern. Ein weiterer
Vorteil ist die Tatsache, dass sich die Aktivierung chemischer Synapsen
auf den Zustand von Nervenzelklen über längere Zeit (bis zu Stunden) auswirken kann.
Synapsen im Nervensystem unterscheiden sich - bei ähnlichem Funktionsprinzip - in einigen Eigenschaften von motorischen Endplatten.
Ähnlich gibt es auch innerhalb des ZNS Unterschiede zwischen speziellen
Synapsentypen. Dies trägt zur Diversität neurophysiologischer Spezifika
im Gehirn bei. Die prinzipiellen Schritte bei der Funktion einer
chemischen Synapse sind die folgenden:

Verpacken des Neurotransmitters in
Speichervesikel, die dann an das präsynaptische Terminal docken
Depolarisierung der präsynaptischen Membran (eintreffendes Aktionspotential)
Öffnung spannungsabhängiger
Calciumkanäle, Einströmen von Ca
++ in das präsynaptische Terminal
Fusion einiger Vesikel mit der präsynaptischen Membran (
Synaptotagmine), Steigerung der Transmitterfreisetzung um einen Faktor
~10
5
Diffusion des Transmitters zur postsynaptischen Membran
Anlagerung einiger Transmittermoleküle an Rezeptoren, postsynaptische Reaktion (Öffnung von Ionenkanälen, Aktivierung von
G-Proteinen,..)
Transmitter wird enzymatisch abgebaut, wieder aufgenommen, oder
abtransportiert

 Abbildung: Synaptische Verschaltungen
Nach einer Vorlage in Carlson NR / Birkett MA, Physiology of Behavior, 12th ed. Pearson 2017
Abbildung: Synaptische Verschaltungen
Nach einer Vorlage in Carlson NR / Birkett MA, Physiology of Behavior, 12th ed. Pearson 2017
Aktionspotentiale entstehen am Axonhügel des Neurons. Von hier werden sie in die Peripherie
geleitet, wo Dendriten, aber auch Nervenkörper (somata) und Axone anderer Neuronen die Signale empfangen und umsetzen
Synapsen ermöglichen neuronale Kommunikation in Millisekunden - über
einen synaptischen Spaltraum hinweg, der 20-40 nm weit sein kann (die Zellmembran hat eine Dicke von ~8 nm).
 Indem er als junger Forscher ein Kapitel für ein Physiologie-Lehrbuch verfasste, führte der britische Neurophysiologe Charles S. Sherrington den
Begriff "Synapse" in die Neurowissenschaften ein. Sherrington, der
später als "Philosoph des Nervensystems" galt, erhielt zusammen mit dem britischen Elektrophysiologen
Edgar D. Adrian
1932 den Nobelpreis für Physiologie oder Medizin "für ihre Entdeckungen
auf dem Gebiet der Funktion der Neuronen". Adrian erforschte vor allem
die Elektrophysiologie von Sinnesorganen.
Indem er als junger Forscher ein Kapitel für ein Physiologie-Lehrbuch verfasste, führte der britische Neurophysiologe Charles S. Sherrington den
Begriff "Synapse" in die Neurowissenschaften ein. Sherrington, der
später als "Philosoph des Nervensystems" galt, erhielt zusammen mit dem britischen Elektrophysiologen
Edgar D. Adrian
1932 den Nobelpreis für Physiologie oder Medizin "für ihre Entdeckungen
auf dem Gebiet der Funktion der Neuronen". Adrian erforschte vor allem
die Elektrophysiologie von Sinnesorganen.
Sind
an einer Synapse die Vesikel rund, die aktive Zone breitflächig, der
synaptische Spalt relativ weit und der die
Signalvermittlung beeinflussende postsynaptische Apparat (postsynaptic density) stark ausgeprägt, spricht man von einer asymmetrischen
Synapse (auch Typ-I-Synapse). Diese ist meist glutamaterg und in der Wirkung exzitatorisch. Typ-I-Synapsen sind meist axo-spinal: Sie finden sich vor allem an Dornenfortsätzen (dendritic spines), an denen man einen Kopf- und einen schlanken Halsteil unterscheiden kann.
Sind die Vesikel abgeflacht, die aktive Zone weniger ausgeprägt, der
synaptische Spalt eher eng und der postsynaptische Apparat zart - die
prä-
und postsynaptische Zone etwa gleich dick -, spricht man von einer symmetrischen
Synapse (auch Typ-II-Synapse). Diese ist meist GABA-erg und in der
Wirkung inhibitorisch. Typ-II-Synapsen finden sich vor allem in
Somanähe (axodendritisch, axosomatisch, axoaxonal).
Elektrische und chemische Synapsen (Abbildung) können interagieren und die "Rechenleistung" neuraler Netzwerke verstärken, z.B. in der Netzhaut, wo neben chemischer Übertragung bipolare und amakrine Zellen auch über gap junctions kommunizieren.
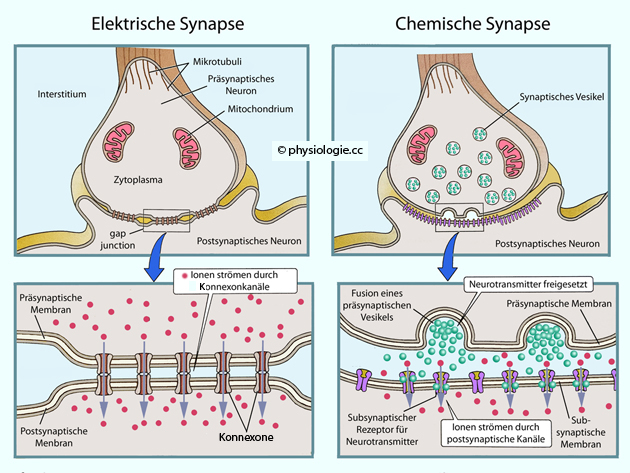
Abbildung: Elektrische vs. chemische Synapse
Nach
einer Vorlage in Augustine / Groh / Huettel / LaMantia / White (eds),
Neuroscience. Intl 7th ed. Oxford University Press 2024
Elektrische Synapsen (links) beinhalten mehrere gap junctions,
durch die zwischen den Zellen Ionen fließen können und damit ein
elektrisches Signal übertragen wird. Bei diesem Synapsentyp sind die
Membranen des prä- und des
postsynaptischen Neurons über Konnexone auf eine Distanz von
weniger als 4 nm angenähert.
Chemische Synapsen (rechts)
haben spezialisierte Anteile - einen präsynaptischen "Sender" und einen
postsynaptischen "Empfänger". Solche Synapsen wirken typischerweise nur
in eine Richtung ("Einbahnverkehr"). Die Membranen sind mindestens 20
nm voneinander entfernt. An der präsynaptischen Membran einer aktivierten Synapse kommt es zur Exozytose des Inhalts präsynaptischer Vesikel (Freisetzung des Neurotransmitters). An der postsynaptischen - insbesondere der subsynaptischen
(unmittelbar vis-a-vis der Exozytosezone liegenden) - Membran binden
Rezeptormoleküle den freigesetzten Neurotransmitter und vermitteln ein
Signal in die postsynaptische Zelle. Der Transmitter kann z.T. zur
präsynaptischen Zelle zurückdiffundieren und von dieser wiederverwertet
werden
Elektrische Synapsen spielen im Gehirn sowohl zwischen Gliazellen - Astrozyten formen Glia-Netzwerke, in denen sich langsame (1-20 µm/s) Ca++-Wellen ausbreiten können - als auch zwischen Neuronen eine Rolle.
Chemische Synapsen setzen an der präsynaptischen Membran bei Erregung
(Aktionspotential) und Calc
ium-Einstrom Transmitterstoffe frei
(mehr als 40 kommen beim Menschen vor; am häufigsten sind
Synapsen glutamaterg).
Sie setzen ihren Transmitter aus synaptischen Vesikeln frei, die in aktiven Zonen eines Axonterminals (synaptic bouton) konzentriert sind. Diese aktiven Zonen sind reich an spannungsgesteuerten Ca++-Kanälen, deren Aktivierung den Exozytosemechanismus startet.
Transmitter wirken auf Rezeptormoleküle
in der "nachgeschalteten" Zelle. Viele dieser Rezeptoren befinden sich
auf Dendritenfortsätzen (eine Purkinje-Zelle in der Kleinhirnrinde hat
z.B. über 2000 Dendriten), und an jedem Dendrit wirken typischerweise mehr
als 100 Synapsen.
Über Dendriten s. dort
Da der gesamte Vorgang komplex ist, benötigt er auch entsprechend Zeit - zwischen
dem präsynaptisch eintreffenden Aktionspotential und der
postsynaptischen Reaktion des Membranpotentials vergeht eine
Verzögerungszeit von meist einer bis einigen Millisekunden (s. Tabelle oben).
Vorgänge am präsynaptischen Apparat:
Der präsynaptische Teil enthält alle Teile für Synthese und
calciumgetriggerte Freisetzung des / der Transmitter(s): Hohe Dichte
von Mitochondrien, um die für den Transportprozess nötige Menge an ATP zu erzeugen; Synthese sowie Speicherung des Transmitters in Vesikeln; spannungsgesteuerte Calciumkanäle zur Aktivierung der Transmitterfreisetzung bei Eintreffen von Aktionspotentialen.
Trifft über das Axon des "sendenden" Neurons ein Aktionspotential ein,
bewirkt dieses die Öffnung von Calc
iumkanälen, und Ca++ dringt in das präsynaptische Axonterminal ein. [Ca++]
in der Nähe der aktiven Zone (hier wird Transmitter freigesetzt) nimmt zu, es fusionieren einige Vesikel
mit der präsynaptischen Membran und geben den Transmitter in den
synaptischen Spaltraum frei ( Abbildung). Dort diffundiert der Transmitterstoff ca. 20 nm zur postsynaptischen Membran.

Abbildung: Sequenz der Signalübermittlung an einer chemischen Synapse
Nach einer Vorlage in Kandel / Koester / Mack / Siegelbaum (eds), Principles of Neural Sciences, 6th ed. 2021 (McGraw Hill)
Der Vorgang erfolgt in mehreren Schritten und erfordert eine Zeit von 0,3-5,0 Millisekunden.
Links: Das Aktionspotential am Axonterminal führt zum Einstrom von Ca++
Mitte: Exozytose führt zur Freisetzzung des Transmitters
Rechts: Der Transmitter bewirkt Na+-Einstrom
und Depolarisierung an den postsynaptischen Rezeptoren (es kann auch
ein anderer Ionenstrom ausgelöst werden, der die Membran hyperpolarisiert)
Das
wiederum löst an Rezeptorkanälen in der postsynaptischen Membran (meist
eines Dendriten, oder auch des Zellkörpers oder Axons) den Einstrom von
Ionen aus, was an der postsynaptischen Zelle zu einer entsprechenden
Änderung des Membranpotentials führt. Der gesamte Vorgang verstärkt das
empfangene Signal.
Was bewirkt der Neurotransmitter an der postsynaptischen Membran?
Das hängt von der Natur des Rezeptors ab, an den er bindet. So kann
Acetylcholin eine Abschwächung (exzitatorische Wirkung, z.B. an
Skelettmuskelzellen) oder auch Verstärkung des Membranpotentials
herorrufen (inhibitorische Wirkung, z.B. an Herzmuskelzellen), je nach
Rezeptor, auf den es trifft.
 Die Aktivierung chemischer Synapsen verändert die Leitfähigkeit ihrer postsynaptischen Membran für monovalente Ionen (Na+, K+, Cl-) und verändert das Membranpotential dementsprechend (Depolarisierung / Hyperpolarisierung).
Die Aktivierung chemischer Synapsen verändert die Leitfähigkeit ihrer postsynaptischen Membran für monovalente Ionen (Na+, K+, Cl-) und verändert das Membranpotential dementsprechend (Depolarisierung / Hyperpolarisierung).
Die Öffnung der postsynaptischen Ionenkanäle durch Bindung des Transmitters kann auf zwei Wegen erfolgen: Einem direkten Gating, bei dem der Rezeptor (an den der Transmitter bindet) identisch ist mit dem Kanal, der die Diffusion der Ionen freigibt; und einem indirekten Gating, bei dem der Rezeptor einen separaten Ionenkanal über den G-Protein-Mechanismus aktiviert ( Abbildung).

Abbildung: Neurotransmitter öffnen postsynaptische Ionenkanäle direkt oder indirekt
Nach einer Vorlage in Kandel / Koester / Mack / Siegelbaum (eds), Principles of Neural Sciences, 6th ed. 2021 (McGraw Hill)
Links:
Der Rezeptor für direktes Gating ist ein Ionenkanal. Bindet der
Transmitter, öffnet sich das "Tor" für Ionen, deren Einströmen in die
Zelle das Membranpotential verändert (ligand-gated channel). Dieser Mechanismus arbeitet sehr rasch.
Rechts: Der Rezeptor für indirektes Gating gehört entweder zur Gruppe der heptahelikalen G-Protein-gekoppelten Rezeptoren (metabotropic receptors),
die ihre Wirkung - in diesem Fall die Aktivierung (Phosphorylierung)
eines Ionenkanals - über zyklisches AMP (cAMP) und Proteinkinase A
(PKA) entfalten (das kann auch Einflüsse auf die Transkription von
Genen und Synthese neuer Proteine einschließen). Oder es sind Rezeptor-Tyrosinkinasen,
welche die Öffnungswahrscheinlichkeit von Ionenkanälen in der Membran
über eine second-messenger-Kaskade beeinflussen (nicht gezeigt)
Die Abbildung zeigt die unterschiedliche Struktur und Wirkungsweise der Rezeptoren: Während das direkte Gating über solche läuft, die auch ein Ionenkanal sind (ionotrop)
und sehr rasche Effekte bewirken (Millisekunden - geeignet für
blitzartige Aktion, z.B. im Rahmen von Muskelspindelreflexen),
funktioniert das indirekte Gating über metabotrope
Rezeptoren - die Ionenkanäle werden indirekt, oft über Proteinkinase A
aktiviert. Letztere haben verzögerte, langsamere und länger wirkende
Effekte zur Folge, z.B. bei Verstärkungsfunktionen im Rahmen von Lernvorgängen.
Abbildung: Synapsen im ZNS
Nach einer Vorlage in Bear / Connors / Paradiso, Neuroscience - Exploring the Brain, 4th ed 2016
Präsynaptische Apparate sind durch aktive Zonen und Vesikel gekennzeichnet (Transmitterfreisetzung), postsynaptische durch Verdichtungszonen (postsynaptic densities)
mit Rezeptoren (Transmitterwirkung).
Die Ausprägung der Synapsen
kann je nach spezieller Funktion sehr unterschiedliche Form annehmen:
Links oben: Axo-spinale Synapse (Dornenfortsatz = dendritic spine)
Rechts oben: Axonaufzweigung, zwei unterschiedlich große präsynaptische Endigungen auf postsynaptischem Soma
Links unten: Große präsynaptische Endigung umfasst postsynaptisches Soma
Rechts unten: Große präsynaptische Endigung wirkt gleichzeitig auf mehrere postsynaptische Kontakte
Es gibt auch dendro-dendritische sowie soma-somatische Synapsen, diese kommen aber selten vor
Die meisten
Synapsen sind axodendritisch, aber es kommen alle möglichen
Kombinationen prä- zu postsynaptischer Verschaltung vor (axosomatisch,
axoaxonal, dendrodendritisch, somatosomatisch, somatodendritisch).
Neurotransmitter
vgl. dort
Neurotransmitter
sind von Nervenzellen - üblicherweise aus Speichervesikeln -
freigesetzte Signalstoffe. Ein Neurotransmitter muss folgende Kriterien
erfüllen, um als solcher qualifiziert zu gelten: (1) Er muss präsynaptisch synthetisiert werden, (2) entsprechende Stimulation des Nerven muss ihn freisetzen, (3) seine synaptische Mikroapplikation muss den Effekt einer Nervenreizung zumindest teilweise nachahmen, und (4) seine Wirkung muss pharmakologisch blockierbar sein.
Neurotransmitter sind mehreren
Stoffklassen zuzuordnen: Aminosäuren, Amine und Neuropeptide. Dazu kommen Cotransmitter wie Purine, NO,
Eikosanoide.
Die wichtigsten Neurotransmitter sind Glutamat (der führende exzitatorische Transmitter) und Aspartat, GABA und Glycin (inhibitorisch), Acetylcholin, Katecholamine, Serotonin und Histamin; als Cotransmitter wirken u.a. Peptide, Cannabinoide, Purine (Adenosin, ATP).
Glycin entsteht aus Serin, GABA und Glutamat aus Glutamin, Aspartat aus
Oxalacetat; Katecholamine aus Tyrosin; Serotonin und Melatonin aus
Tryptophan, Histamin aus Histidin; Acetylcholin
aus Cholin; Cannabinoide aus Phospholipiden; NO aus Arginin. Solche
kleinen Transmitter können im Prinzip überall im Neuron entstehen, wo
entsprechende Enzyme vorhanden sind.
In den meisten Fällen sind Neurotransmitter Aminosäuren (Glutamat und Aspartat wirken erregend, Glycin und GABA hemmend), einfache Amine (Acetylcholin, Serotonin, Noradrenalin..) oder Peptide (z.B. Tropine des Hypophysenvorderlappens). Auch Purine (Adenosin, ATP), Endocannabinoide, oder Gase (NO, CO, H2S) kommen als Transmitter in Frage.
Entgegen der klassischen Sicht können Neurone auch mehr als einen Neurotransmitter freisetzen: Häufig werden an ein un derselben Snapse mehrere Transmitterklassen gespeichert (Colokalisierung) und freigesetzt (Cotransmission, co-release) - ob aus unterschiedlichen oder identischen Vesikeln, ist nicht immer klar.
Ob Transmitter exzitatorisch (depolarisierend), inhibitorisch
(hyperpolarisierend) oder als Neuromodulatoren wirken, hängt vor allem
von den Rezeptoren ab, auf die
sie postsynaptisch treffen. So wirken Glutamat oder Acetylcholin im
Allgemeinen erregend, in bestimmten Situationen aber auch hemmend auf
postsynaptische Zellen.
Häufig vorkommende Neurotransmitter und ihre Eigenschaften

Kombiniert / teilweise modifiziert nach Augustine / Groh / Huettel / LaMantia / White (eds),
Neuroscience. Intl 7th ed. Oxford University Press 2024; und Liqun Luo, Principles of Neurobiology, 2nd ed. CRC Press 2021 (erweitert)
|
|
Neuro-
transmitter
|
Vorkommen
|
Cotransmission
| Vorläufer
|
Postsynapt. Effekt
|
Synthese- limitierender Schritt
|
Entfernungs-
mechanis-
mus
|
Vesikeltyp
|
Acetylcholin
|
Motorische Neurone zu Muskeln; Neurone im autonomen Nervensystem; exzitatorische / modulatorische Neurone im ZNS
|
VIP
Substanz P
| Cholin + Acetyl-CoA
|
E
|
ChAT
(Cholin Acetyl-
transferase)
|
AChE (Acetyl- cholinesterase)
|
klein
|
Glutamat
|
Mehrzahl der exzitatorischen Neurone im ZNS; meiste sensorische Neurone
|
| Glutamin
|
E
|
Glutaminase
|
Transporter
|
klein
|
GABA
|
Meiste inhibitorische Neurone im ZNS |
Somatostatin
Cholecystokinin
Neuropeptid Y
| Glutamat
|
I
|
GAD
(Glutamat-
Decarboxylase)
|
Transporter |
klein
|
Glycin
|
Einige inhibitorische Neurone (hauptsächlich in Hirnstamm und Rückenmark)
|
| Serin
|
I
|
Phosphoserin
|
Transporter |
klein
|
Serotonin (5-HT)
|
Modulatorische Neurone im ZNS; Neurone im Gastrointestinaltrakt
|
Substanz P
TRH
Enkephaline
| Tryptophan
|
I
|
Tryptophan-
hydroxylase
|
Transporter
MAO
|
groß
dichter Kern
|
Dopamin
|
Modulatorische Neurone im ZNS |
Cholecystokinin
Neurotensin
GLP-1
| Tyrosin
|
E
|
Tyrosin-
hydroxylase
|
Transporter
MAO, COMT
|
klein, dichter Kern, oder groß, heterogen
|
Noradrenalin
|
Modulatorische Neurone im ZNS; autonom-nervöse Neurone
|
Galanin
Enkephaline
Neuropeptid Y
| Tyrosin
|
E
|
Tyrosin-
hydroxylase
|
Transporter
MAO, COMT |
klein, dichter Kern, oder groß, heterogen |
Histamin
|
Modulatorische Neurone im ZNS |
| Histidin
|
E
|
Histidin-
decarboxylase
|
Transporter
|
groß, dichter Kern
|
ATP, Adenosin
|
Einige sensorische und ZNS-Neurone
|
| ADP
|
E
|
Mitochondrielle oxidative Phoyphory-
lierung, Glykolyse
|
Hydrolyse zu AMP + Adenosin
|
klein
|
Neuropeptide
|
Exzitatorische, inhibitorische, modulatorische Neurone (Cotransmission);
neurosekretorische Zellen
|
Mit Katecholaminen, Acetylcholin, GABA, Serotonin, Oxytocin, Vasopressin
| Aminosäuren
(Protein-
synthese)
|
E + I
|
Synthese und Transport
|
Proteasen
|
groß, dichter Kern |
Endo-
cannabonoide
|
ZNS, peripheres Nervensystem, Immunsystem, Organe
|
Neurotransmitter
|
Membran-
lipide
|
hemmen Freisetzung z.B. von GABA
|
Enzymatische Modifikation von Lipiden
|
Hydrolyse durch FAAH (Fettsäureamid- hydrolase)
|
--
|
Stickstiff-
monoxid (NO)
|
Endothel
|
CO, VIP, ATP
|
Arginin
|
E + I
|
NO-Synthase
|
Spontane Oxidation
|
--
|
Vesikelzyklus, Speicherung, Freisetzung und Recycling von Neurotransmittern
Präsynaptische Vesikel
entstehen zunächst aus Abschnürungen aus dem Golgi-Apparat (dieser
verbringt die im rauen endoplasmatischen Retikulum synthetisierten
Proteine sekretorische Vesikel). Die Vesikel haben in ihrer Wand Protonenpumpen (aktiver Transport), die den pH-Wert auf ~5,4 einstellen. Der Protonengradient zum Zytoplasma (pH 7,2) treibt Antiporter
an, die Transmitter im Vesikel konzentrieren (sekundär aktiver
Transport). Auch der elektrische Gradient kann für den
Transmittertransport in die Speichervesikel genutzt werden.
Die Speicherung von Neurotransmittern in Vesikeln hat mehrere Vorteile:
Anreicherung bis zum 105-fachen der Konzentration im
Zytoplasma,
Schutz vor Abbau,
Reserve für die synaptische
Aktivität.
Wie
gelangen die im Soma der Nervenzelle gebildeten Vesikel (auch
Mitochondrien) in die Peripherie (das Axon-Terminal kann bis zu ca.
einen Meter entfernt sein)? Diese Aufgabe übernehmen Mikrotubuli im
Axonfortsatz (schneller axonaler Transport bewältigt 0,4 m in 24 Stunden).
Nervenzellen
bilden Neurotransmitter frisch oder recyceln diese durch Wiederaufnahme
aus dem perisynaptischen Extrazellulärraum; die Transmitterstoffe
werden dann in Vesikeln gespeichert, um bei Erregung des Axons in den
synaptischen Spaltraum abgegeben zu werden. Neuropeptide werden zunächst im Golgi-Apparat durch
post-translationale Prozessierung fertiggestellt; dann sprossen mit fertigem Transmitter gefüllte Vesikel aus und werden in die
Peripherie des Axons transportiert.
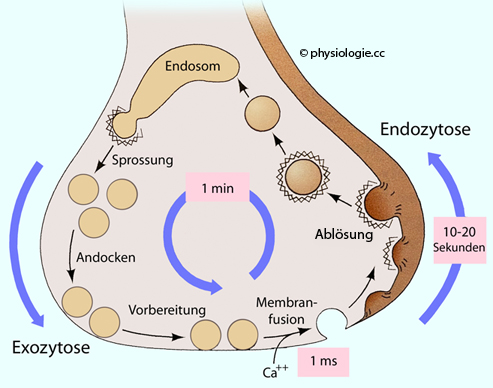
Abbildung: Recycling synaptischer Vesikel
Nach
einer Vorlage in Augustine / Groh / Huettel / LaMantia / White (eds),
Neuroscience. Intl 7th ed. Oxford University Press 2024
Der
gesamte Zyklus dauert etwa eine Minute: Mit Transmittermolekülen
beladene synaptische Vesikel sprossen aus dem endosomalen Kompartiment
ab, begeben sich zur präsynaptischen Membran, fusionieren mit dieser (calciuminduziert) mit Hilfe des SNARE-Systems und exozytieren ihren Inhalt. Nach der Fusion wird das (mit Proteinen bedeckte) Membranmaterial endozytiert und wiederverwertet
Ein Vesikelzyklus
(lokales Recycling, bestehend aus budding - Aussprossen ("Knospung") eines Vesikels, docking - Anlagerung an die
präsynaptische Membran, priming - Vorbereitung, fusion - Verschmelzen
mit der präsynaptischen Membran, budding - endozytotische Abspaltung von der Zellmembran, Rückführung über Golgi-Apparat / Endosomen) dauert etwa eine Minute (Abbildung), die Membranfusion und Freisetzung des Neurotransmitters lediglich eine Millisekunde.
Präsynaptische Vesikel sind Angriffspunkt für negative
Rückkopplung. Beispiele: Bei starker Freisetzung des Transmitters nimmt ihre
Synthese zu, bei geringer präsynaptischer Aktivität hingegen ab. Synapsin, das reversibel an Vesikel bindet, verhindert deren Freigabe; CaMKII phosphoryliert Synapsin und
bewirkt seine Ablösung vom Vesikelpool, was das Andocken einzelner Vesikel an die
präsynaptische Membran und folglich die Freigabe des Neurotransmitters ermöglicht (s. weiter unten).
Synapsine sind
eine Proteinfamilie (Synapsin I, II, III), die vor allem von
Nervenzellen exprimiert werden und präsynaptische Vesikel untereinander
und mit dem Zytoskelett zu einem "Reservepool" verbinden und in diesem
fixieren. Dadurch beeinflussen sie die Verfügbarkeit der Vesikel für
die synaptische Freigabe der in ihnen gespeicherten Neurotransmitter.
Bei Erregung der Zelle (Aktionspotentiale) werden Synapsine durch
Proteinkinasen phosphoryliert, was sie aus dem Netzwerk mit anderen
Vesikeln löst und die Freisetzung des Transmitters in den synaptischen
Spaltraum (Exozytose) ermöglicht.
CaMKII - CaM Kinase II, Ca++/calmodulin dependent protein kinase II - ist eine Proteinkinase, die durch den Ca++ / Calmodulin-
(CaM-) Komplex gesteuert wird. Sie ist an zahlreichen zellulären
Vorgängen beteiligt, u.a. der Mobilisierung präsynaptischer Vesikel (s.
oben), der Wiederaufnahme von Calciumionen in Herzmuskelzellen, an
Lern- und Gedächtnisprozessen (synaptische Langzeitpotenzierung), an
der T-Zell-Aktivierung, oder als Myosin-Leichtkettenkinase.
Autophosphorylierung kann CaMKII dauerhaft aktiv machen.
Man
unterscheidet kleine und große präsynaptische Speichervesikel. Beide
enthalten ATP, das in verschiedener Weise genutzt werden kann.
Die Mehrzahl der präsynaptischen Vesikel hat einen Durchmesser von ~40 nm,
sie speichern kleine Nichtpeptide wie Glutamat, GABA, Acetylcholin. Diese Vesikel finden sich in der Nähe von aktiven Zonen und erscheinen
elektronenmikroskopisch "leer" (clear / small vesicles). Ihre Wirkung beschränkt sich auf klar begrenzte synaptische Strukturen.
Einige Vesikel sind größer (70-250 nm Durchmesser: large dense-core vesicles,
homolog sekretorischen Granula von Nicht-Nervenzellen), gleichen sekretorischen Granula in endokrinen Zellen (dense-core secretory granules) und enthalten Neuropeptide, die aus Vorstufen synthetisiert und zusammen mit anderen Proteinen in das
Trans-Golgi-Netzwerk gebracht werden. Aus diesem separieren sich Vesikel, und diese werden vom Soma zu
präsynaptischen Zielen transportiert. Kleinmolekulare Transmitter und andere neuroaktive Moleküle (Cotransmitter) können zusammen mit dem Neuropeptid in dense-core-Vesikeln gespeichert und zusammen mit diesem freigesetzt werden.
Solche Vesikel sind über das ganze präsynaptische Endstück des Neurons verteilt; ihr Inhalt kann überall am Neuron
exozytiert werden, ausgelöst durch Aktionspotentialsalven, Öffnung
spannungssensitiver Ca++-Kanäle und dadurch Erhöhung der intrazellulären [Ca++]. Im Gegensatz zu kleinen synaptischen Vesikeln wird ihre
Membran nicht recycelt. Für freigesetzte Neuropeptide gibt es keinen
Reuptake-Mechanismus, und sie müssen im Soma der Zelle neu
synthetisiert und mittels anterograden Transports in die Peripherie gebracht werden.
Freisetzung des Transmitters aus präsynaptischen Vesikeln
Aktionspotentiale (Depolarisierung) öffnen Ca++-Kanäle, Calc
iumionen lösen die Exozytose (Priming - Docking - Fusion) des
Transmitters aus ( Abbildung).
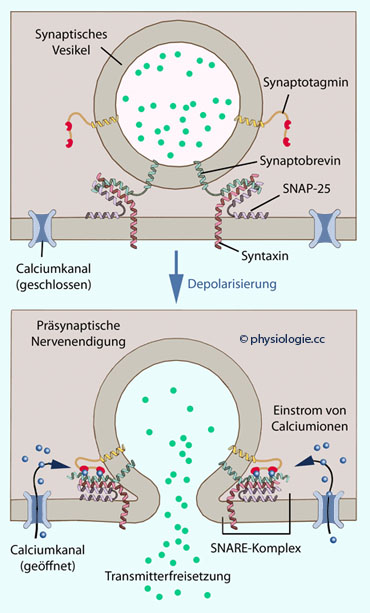
Abbildung: Mechanismus der Neurotransmitterfreigabe aus präsynaptischen Vesikeln
Nach einer Vorlage bei Guyton and Hall, Textbook of Medical Physiology, 15th ed. Elsevier 2026
Damit
sich präsynaptische Vesikel zum synaptischen Spalt hin öffnen
(Membranfusion) und den in ihnen gespeicherten Transmitter freisetzen
können, müssen Calciumionen aus der extrazellulären Flüssigkeit in das
Zytoplasma gelangen. Das geschieht durch Depolarisierung der
Zellmembran (Ankunft von Aktionspotentialen); darauf reagieren spannungssensitive Calciumkanäle, die sich öffnen, und Calciumionen binden an Membranproteine (Synaptotagmin, Synaptobrevin).
Dadurch bilden sich Molekülbrücken in einem Komplex membrangebundener Proteine, der als SNARE (Soluble NSF Attachment Protein Receptor)
bezeichnet wird. So wird die Fusion der Membran ermöglicht, und der
Inhalt der Vesikel kann in den synaptischen Spaltraum diffundieren
Die Freisetzung aus den Vesikeln erfolgt nach einem generellen Schema: Depolarisierung (Na+-Einstrom) öffnet spannungsgesteuerte Ca++-Kanäle (vor allem vom N-Typ und P-Typ), wobei die Dauer des Aktionspotentials die Intensität des Ca++-Einstroms bestimmt. Bei Erregung des Neuriten kommt es zur Exozytose von 50-100 Vesikeln - das heißt, jedes Aktionspotential führt zur Freisetzung eines
beträchtlichen Anteils des präsynaptisch gespeicherten
Neurotransmitters in den synaptischen Spaltraum.
Synaptische Vesikel tragen in ihrer Membran bzw. an diese angeheftet verschiedene Proteine (SNARE-Proteine: Soluble NSF Attachment Protein Receptor - NSF: N-ethylmaleimide-sensitive factor, N-ethylmaleimide sensitive fusion protein, Abbildung
en).
Man unterscheidet
 v-SNAREs in der Wand von Vesikeln (v = vesicle) und
v-SNAREs in der Wand von Vesikeln (v = vesicle) und
t-SNAREs (t = target) in der präsynaptischen Membran.
An der Fusion transmitterspeichernder Vesikel mit der präsynaptischen Membran sind SNARE-Komplexe beteiligt
|
Das "Trafficking"
präsynaptischer Vesikel erfordert zahlreiche spezialisierte Proteine;
die zytoplasmatische Seite der Vesikelmembranen sind dicht mit ihnen
besetzt. Zu solchen Proteinen zählen
 Synapsin, das reversibel an Vesikel bindet und diese wahrscheinlich untereinander zu einem Netzwerk verknüpft, was einen stabilen
Reservepool an transmittergefüllten Vesikeln ergibt. Phosphorylierung
von Synapsin durch Proteinkinasen (vor allem CaMKII, Ca2+/calmodulin-dependent protein kinase, type II)
bewirkt die Ablösung des Synapsins von Vesikeln, was diese für eine
Interaktion mit SNARE-Proteinen freigibt und das Andocken an die
präsynaptische Membran ermöglicht.
Synapsin, das reversibel an Vesikel bindet und diese wahrscheinlich untereinander zu einem Netzwerk verknüpft, was einen stabilen
Reservepool an transmittergefüllten Vesikeln ergibt. Phosphorylierung
von Synapsin durch Proteinkinasen (vor allem CaMKII, Ca2+/calmodulin-dependent protein kinase, type II)
bewirkt die Ablösung des Synapsins von Vesikeln, was diese für eine
Interaktion mit SNARE-Proteinen freigibt und das Andocken an die
präsynaptische Membran ermöglicht.
Syntaxine
und SNAP-25 sind Bestandteile der Zellmembran. Sie formen zusammen einen makromolekularen Komplex, der die beiden (Zell- und Vesikel-) Membranen nahe zusammenbringt. Syntaxine können Synaptotagmine und Synaptobrevine
binden und wahrscheinlich einen Teil der exozytotischen
Fusionsporen bilden. SNAP-25 (SNAp REceptors - SNAP = synaptosomal-associated protein),
das bei Aktivierung (durch allosterische Veränderung des
Synaptotagmins) zusammen mit Synaptobrevin und Syntaxin den
SNARE-Komplex bildet, spielt bei der Membranfusion / Exozytose eine essentielle Rolle.
Synaptotagmine (SYTs) sind vesikelgebundene Proteine, die Ca++ binden können. Strömen Calciumionen infolge einer Depolarisierung der Membran in die Zelle, wirken sie als intrazellulärer Ca++-Sensor und lösen die Exozytose und Freisetzung des Transmitters aus. Im Ruhezustand (niedrige zytoplasmatische
Calciumkonzentration) verhindern sie die Verschmelzung von Vesikeln mit der
präsynaptischen Membran.
Das SNARE-Protein
Synaptobrevin
(Isotypen 1 und 2) ist als Bestandteil der Vesikelmembran ein
Schlüsselprotein für die Membranfusion. Es beteiligt sich an der Fusion
synaptischer Vesikel mit der präsynaptischen Membran im Rahmen der Exozytose.
Insgesamt kennt man beim Menschen ≥35 verschiedene SNARE-Proteine.

Abbildung: Organisation einer präsynaptischen Endigung
(presynaptic terminal)
Nach einer Vorlage in Liqun Luo, Principles of Neurobiology, 2nd ed. CRC Press 2021
Links:
Transsynaptische Adhäsionsmoleküle (wie Cadherin-Cadherin: homophile Bindung, Neurexin-Neuroligin: heterophile Bindung, vgl. dort)
halten die aktive Zone der präsynaptischen Endigung auf einem
definierten Abstand zur Membran der postsynaptischen Membran. Deren
hohe Konzentration an Rezeptoren erhöht die Wirkgeschwindigkeit
freigesetzten Neurotransmitters.
Rechts: Vergrößerter Ausschnitt
des präsynaptischen Teils. Unc13 bindet SNAREs und nähert synaptische
Vesikel an die präsynaptische Membran an. Der RIM/RIM-BP-Komplex bindet
einerseits direkt an spannungssensitive Calciumkanäle, andererseits
über Rab3 an synaptische Vesikel. So führt Einstrom von Calciumionen
direkt zur Aktivierung von Synaptotagmin. Dies wiederum hebt den durch
Complexin bdeingten Block des SNARE/Munc18-Komplexes auf und ermöglicht
die Freisetzung des Neurotransmitters. RIM und RIM-BP sind mit
Gerüstproteinen der Zelle verknüpft
Aktive Zonen (Abbildung
) sind
Teile der präsynaptischen Membran, mit denen Vesikel fusionieren und
hier ihren Inhalt (Transmitter) in den synaptischen Spaltraum
freisetzen. Sie liegen rezeptorbeladenen postsynaptischen
Membranflächen direkt gegenüber. Die meisten Synapsen im ZNS weisen nur
wenige aktive Zonen auf (oft nur eine, manchmal bis zu 20).
Trifft ein Aktionspotential an einer Synapse ein, bedeutet das nicht,
dass diese auch Transmitter freigibt: Vielmehr besteht dafür nur eine
bestimmte Wahrscheinlichkeit (release probability), die meist deutlich unter 1 (100%) liegt. Die Wahrscheinlichkeit der Exozytose von Transmitter steigt mit Zahl an aktiven Zonen zwischen der präsynaptischen und der postsynaptischen Zelle. Sie hängt weiters von der vonausgegangenen synaptischen Aktivität ab: Bahnende Synapsen (facilitating synapses) erhöhen die Stärke der postsynaptischen Reaktion auf präsynaptische Erregung, hemmende (depressing synapses)
senken sie ab. Ob die Synapse bahnt oder hemmt, hängt auch von der
unmittelbaren Vorgeschichte der Erregungsgröße ab; so kann sich bei
wiederholter Reizung (Aktionspotentialsalven) zunächst eine
"Calciumwolke" im Bereich der aktiven Zone aufbauen und die
Wahrscheinlichkeit der Transmitterfreisetzung steigern, später kann es
durch Erschöpfung des Vesikelpools zum gegenteiligen Effekt kommen.
Das Protein Unc13 (Abbildung)
spielt eine zentrale Rolle bei dem Mechanismus, der die Exozytose
organisiert: Es bindet und aktiviert t-SNAREs (Membranproteine; t = target)
und fixiert gleichzeitig das v-SNARE (v = vesicle) Synaptobrevin an die
Stelle, wo der Transmitter in den synaptischen Spalt abgegeben werden
soll. Zwei weitere Komponenten der aktiven Zone sind RIM (Rab3-interacting molecule) und RIM-BP (RIM-bindung protein).
RIM bindet die GTPase Rab3 und befördert synaptische Vesikel in die
Nähe spannungssensitiver Calciumkanäle der präsynaptischen
Membran.
Rab3 ist eine Familie kleiner GTP-asen (Rab3A, 3B, 3C, 3D), die beim vesicle trafficking als molekulare Schalter fungieren. Der Name leitet sich von Ras-associated binding ab.
Der RIM/RIM-BP-Komplex interagiert auch mit weiteren Proteinen der
aktiven Zone, die ihrerseits mit dem Zytoskelett (Actinfäden)
interagieren; so ist ein Konnex zum anterograden Molekültransport vom Soma des Neuriten zur präsynaptischen Peripherie gegeben.
SNARE-Proteine beschleunigen die Fusion von Vesikeln mit der Zellmembran - das "Andocken" von Vesikeln an
die präsynaptische Membran und die Ausbildung von Poren in der
Vesikelwand
im Rahmen der Fusion von Vesikel- und Axonmembran - sie "entsperren"
den Transmitter, der in Vesikeln gespeichert vorliegt.
Die Aufnahme des Transmitters durch vesikuläre Transmittertransporter wird durch einen niedrigen vesikulären pH-Wert angetrieben, der durch H+-Pumpen aufrechterhalten wird.
 Neurotoxine hemmen die Exozytose (vor allem an der motorischen Endplatte) durch Spaltung von SNARE-Proteinen (Botulinumtoxine A und E → SNAP-25, Botulinumtoxin B → Synaptobrevin). Das Clostridiengift Botulinumtoxin (Botox) kann therapeutisch verwendet werden, z.B. um Muskelkrämpfen gegenzuwirken (i.m. Injektion bei Spasmen).
Neurotoxine hemmen die Exozytose (vor allem an der motorischen Endplatte) durch Spaltung von SNARE-Proteinen (Botulinumtoxine A und E → SNAP-25, Botulinumtoxin B → Synaptobrevin). Das Clostridiengift Botulinumtoxin (Botox) kann therapeutisch verwendet werden, z.B. um Muskelkrämpfen gegenzuwirken (i.m. Injektion bei Spasmen).
Die präsynaptische Freisetzung von Acetylcholin wird durch Botulinumtoxin spezifisch gehemmt
|
Auch Tetanustoxin
spaltet SNAREs - genauer: Synaptobrevin - und verhindert dadurch die
Freisetzung von inhibitorischen Neurotransmittern (Glycin und GABA).
Das betrifft die Selbsthemmung der motorischen Vorderhornzellen durch
Renshaw-Zellen, und sie werden übererregbar - es treten Muskelkrämpfe
auf.
Tetanustoxin spaltet Synaptobrevin und verhindert die Glycin-Freisetzung an Renshaw-Zellen
|
Jedes
Aktionspotential führt zur Entleerung von einigen hundert Vesikeln, was
die Freisetzung von einigen zehntausend Transmittermolekülen bedeutet.
Der freigesetzte Transmitter wird anschließend z.T. wiederaufgenommen,
z.T. wird neu synthetisierter Transmitter aus dem Soma nachgeliefert (axonaler Transport).
Mehrere Faktoren können präsynaptisch die Menge des freigesetzten
Transmitters (pro Aktionspotential) und damit die Intensität der
synaptischen Signalübertragung variieren (synaptische Plastizität): Einerseits über den Calciumeinstrom (dieser kann durch präsynaptisch wirkende - axoaxonal freigesetzte - Neuromodulatoren
beeinflusst werden; wiederholte Reizung kann den Calc
iumeinstrom
verstärken - Fazilitation - oder abschwächen - Depression),
andererseits durch die Reaktionsstärke der Vesikelfusion pro gegebener Calc
iummenge.
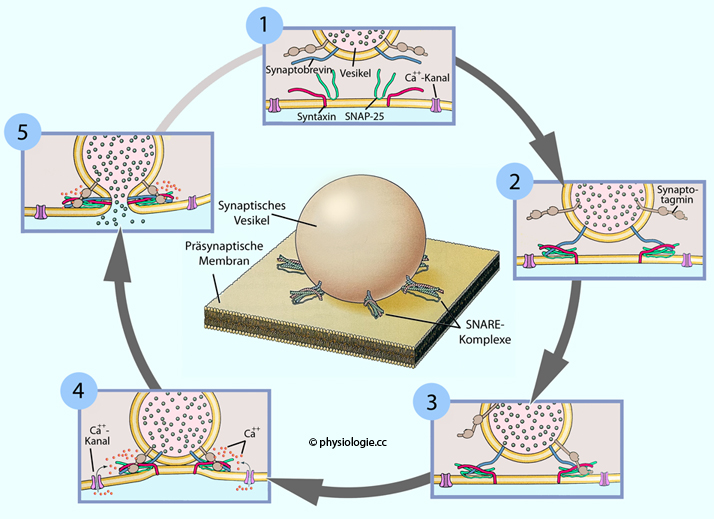
Abbildung: Modell des SNARE-Mechanismus bei synaptischer Vesikelfusion
Modifiziert nach
einer Vorlage in Augustine / Groh / Huettel / LaMantia / White (eds),
Neuroscience. Intl 7th ed. Oxford University Press 2024
In
der Bildmitte ist die Organisation der SNARE-Komplexe eines angedockten
Vesikels gezeigt: Sechs solche Komplexe sind symmetrisch um die
Berührungszone des Vesikels mit der präsynaptischen Membran angeordnet.
Die Boxen (1-5) zeigen ein Modell der calciumgetriggerten
Vesikelfusion: Während des Andockens eines synaptischen Vesikels bauen
SNARE-Proteine (1: Synaptobrevin und Synaptotagmin aus der Membran des
Vesikels, Syntaxin und SNAP-25 aus der präsynaptischen Zellmembran)
einen Proteinkomplex auf, der die Membranen zusammenbringt.
2: Beginn der Bildung eines SNARE-Komplexes, 3: Synaptotagmin bindet an den SNARE-Komplex.
4: Calciumionen strömen durch Calciumkanäle (die infolge der Depolarisierung geöffnet sind) in das
präsynaptische Terminal und binden an Synaptotagmin, das sich -
solchermaßen getriggert - in der präsynaptischen Membran verankert
und die Öffnung einer Pore ermöglicht, die sich weitet und den Austritt
des Neurotransmitters bewirkt (5)
Die Fusion der synaptischen Vesikel mit der präsynaptischen Membran involviert Syntaxin und Chaperone. R-SNAREs wirken als v-SNAREs, Q-SNAREs als t-SNAREs. Expandiert die Fusionspore bei der Freisetzung des Transmitters, werden Trans- zu Cis-SNARE-Komplexen.
Die Öffnung der Ca++-Kanäle erfolgt langsamer als die der Na+-Kanäle und kostet 1-2 ms Zeit; Ca++-Ionen
strömen erst dann in das präsynaptische Terminal ein, wenn das
auslösende Aktionspotential schon weitgehend abgelaufen ist (synaptic delay).
Calc
iumionen bewirken die Exozytose des Neurotransmitters (elektro-sekretorische Kopplung).
Dies ist ein Vorgang von erheblicher Komplexität (SNARE-Zyklus s. Abbildung). Ca++-Ionen aktivieren über Calmodulin und eine Proteinkinase aktiviert
Synapsine - mit dem Zytoskelett
verbundene Membranproteine, welche die Stabilität der Vesikel steuern
-, was weitere Vesikel für die Transmitterfreigabe vorbereitet.
Die
Fusion der Vesikelmembran mit der präsynaptischen Zellmembran dauert
nur Bruchteile einer Millisekunde. Bis der Transmitter dann aus den
Vesikeln in den synaptischen Spalt freigesetzt ist, braucht es
Bruchteile einer Sekunde.
Recycling durch präsynaptische Endozytose
Für eine verlässliche Synapsenfunktion - insbesondere bei
hochfrequenten Aktionspotentialfolgen - ist die Wiederverwertung von
synaptischen Vesikeln essentiell. Daher gibt es einen sparsamen Umgang
mit diesen Transmitterbehältern, da die Membranbestandteile und
involvierten Proteine nicht vor Ort, sondern im Soma der Nervenzelle
synthetisiert und in die Peripherie transportiert werden müssen. Für
dieses Recycling kommen mehrere Mechanismen in Frage:

Abbildung: Zyklus der synaptischen Vesikel
Nach einer Vorlage in Liqun Luo, Principles of Neurobiology, 2nd ed. CRC Press 2021
Nach der Freisetzung des Transmitters können synaptische Vesikel auf drei Wegen wiederverwendet werden:
1a, Kiss-and-run:
Nach einer kurzen Öffnung zum Synapsenspalt mit limitiertem Austausch
von Lipid- und Proteinmolekülen mit der präsynaptischen Membran bilden
sich die Vesikel rasch wieder zurück.
1b, Clathrinmediierte Endozytose:
Die Vesikelmembran fusioniert vollständig mit der präsynaptischen
Membran und wird anschließend mittels des Clathrinapparats wieder
zurückgewonnen.
1c, Ultraschnelle Endozytose: Die präsynaptische Membran wird rasch endozytiert, dabei entstehen große Vesikel, die über einen Clathrinmechanismus mit
Endosomen fusionieren. Protonenpumpen säuern anschließend den Inhalt
der Vesikel an, die dann mit Neurotransmitter befüllt werden (2) und sich in den präsynaptischen Vesikelpool einfügen (3). Einige devon rücken zur aktiven Zone vor (4) und fusionieren abermals mit der Membran (5)
Einige Vesikel können sich nach ihrer Entleerung - statt mit der
präsynaptischen Membran zu fusionieren - auch wieder rekonfigurieren
und in das präsynaptische Zellinnere zurückziehen ("kiss and run"-Mechanismus, 1a in der Abbildung).
Dabei
wird nur ein Teil der im Vesikel gespeicherten Transmittermenge an den
Extrazellulärraum freigegeben.
Dies
spart Stoffwechselenergie, weil das Vesikel mehrfach
hintereinander zum Einsatz kommt, ohne remodifiziert zu werden. Vor
allem geht nur ein kleiner Anteil der Bestandteile der
Vesikelmembran an die präsynaptische Zellmembran verloren.
Eine andere Möglichkeit ist eine durch Clathrin ( Abbildung) vermittelte
Rückgewinnung der Membran nach ihrer Fusionierung mit der Zellmembran;
sie dauert mehrere Sekunden (1b in der Abbildung oben). Schließlich gibt es auch einen sehr
raschen (etwa eine Zehntelsekunde dauernden) Endozytosemechanismus, bei
dem sich aus der die aktive Zone begrenzenden präsynaptischen Membran
große Vesikel bilden und dann über einen Clathrinmechanismus zu
großen synaptischen Endosomen werden, die zur Transmitterspeicherung verwendet werden (1c).
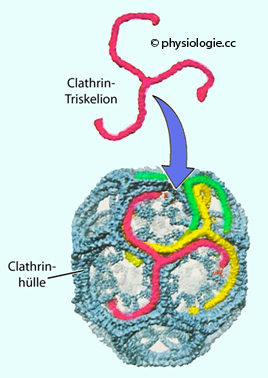 Abbildung: Clathrin und endozytotisches Vesikel
Abbildung: Clathrin und endozytotisches Vesikel
Nach
einer Vorlage in Augustine / Groh / Huettel / LaMantia / White (eds),
Neuroscience. Intl 7th ed. Oxford University Press 2024
Clathrinmoleküle
ordnen sich an der Membran zu gitterförmigen Strukturen an und zwingen
ihr aufgrund der gekrümmten Molekülgestalt insgesamt eine kuppelartige
Form auf. Mehrere Adapterproteine (AP-2, AP-180, Amphiphysin, Epsin
etc) befestigen die Clathrinschicht an Proteinen und Lipiden der
Membran.
Die Clathrinhülle lässt kugelförmige Strukturen entstehen, die als clathrin coated pits
bezeichnet werden und die Grundlage bilden für weitere Abschnürung von
der Außenmembran und die Ausbildung endozytotischer Vesikel (folgende
Abbildung).
Clathrin ist das wichtigste Protein für die Endozytose. Es hat eine besondere Struktur mit drei gebogenen Fortsätzen (Triskele, triskelion ), die an ihren C-Enden gekoppelt sind.
Die sternförmigen Moleküle können sich so zusammenfügen, dass sich eine
kuppelartige Struktur ergibt, welche die Membran zusehends aufwölbt und
letztlich ihr Ablösen aus der Zellmembran und die Bildung endozytotischer Vesikel ermöglicht.
Zunächst ist der entstandene kugelförmige Clathrinkäfig noch mittels
eines Lipidstiels (16 nm Durchmesser) an der Zellmembran befestigt. Um
diesen formiert sich eine spiralförmige Proteinstruktur
(12 nm Abstand der Spiralarme), bestehend aus Dynamin (Abbildung). Diese Umklammerung bewirkt die
endgültige Trennung des coated pit von der Zellmembran und die (durch
Aktinfilamente vermittelte) Wanderung des nunmehrigen clathrin coated vesicle in das Zytoplasma.
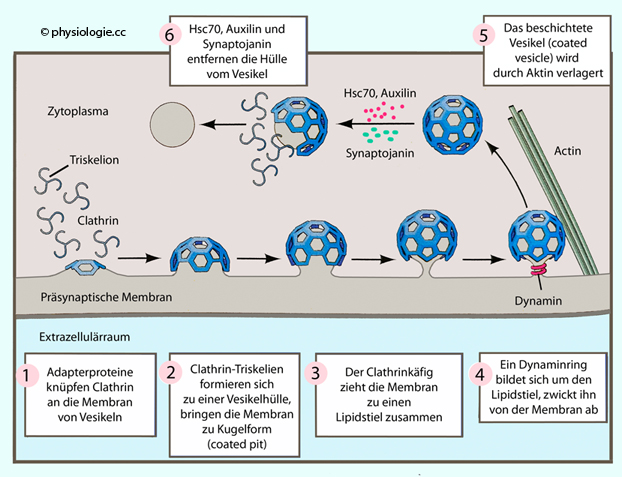
Abbildung: Modell der Membranablösung im Rahmen einer Endozytose
Nach
einer Vorlage in Augustine / Groh / Huettel / LaMantia / White (eds),
Neuroscience. Intl 7th ed. Oxford University Press 2024
Clathrinmoleküle (Triskelien) lagern sich an der Innenseite der präsynamptischen Membran an, es entstehen "Membrandome" (coated pits).
Diese wachsen zu kugelförmigen Strukturen weiter, die schließlich nur
noch über einen dünnen Lipidstiel an der Zellmembran befestigt sind. An
diesen lagern sich Dynaminmoleküle an, welche die endgültige Trennung von der Membran ermöglichen.
Die nunmehr von der Zellmembran unabhängigen coated vesicles
bewegen sich via Actinfilamente in das Zellinnere. Unter der Wirkung
mehrerer Enzyme (wie der Phosphatase Auxilin, der ATPase
Hitzeschockprotein Hsc70, der Lipidphosphatase Synaptojanin) fällt der
Clathrinkäfig vom Vesikel ab, und dieses kann den Vorgang des
Recyclings - Ansäuerung des Inhalts über Protonenpumpen (vATPasen) zwecks Befüllen mit Neurotransmitter, Wiederverwendung bei Erregung des Neurons) - fortsetzen
Die
Polymerisierung des Clathrinkäfigs erfolgt im Zusammenwirken mit dem an
der Membraninnenseite liegenden Adapterprotein Amphiphysin,
das
verschiedene Bindungsstellen für Membrankomponenten aufweist und im
Rahmen der Vesikelabspaltung Dynaminmoleküle rekrutiert. Amphiphysin
wirkt nicht nur in Nerven-, sondern auch in Muskelzellen, wo es sich an
der Bildung des T-tubulären Netzwerks beteiligt.
Der synaptische
Spaltraum ist 20-30 nm weit; er ist Teil des extrazellulären Raums. Die sogenannte aktive Zone der synaptischen Membranen verfügt über
zahlreiche verschiedene Adhäsionsmoleküle, welche die präsynaptische an die
postsynaptische Membran anheften. Das gewährleistet einen
konstanten und geringen Abstand für die Diffusion
freigesetzter Transmittermoleküle zu den postsynaptischen Rezeptoren
und möglichst rasche Neurotransmission.
Zu den makromolekularen Komplexen zählen (präsynaptisch) Neurexine in diversen Ausführungen (alternatives Splicing), die an verschiedene (postsynaptische) Neurexinrezeptoren - wie Neuroligin - binden (heterophile Bindung, Abbildung).
Cadherine binden an Cadherin (homophile Bindung).

Abbildung: Synaptische Molekülnetze
Nach Feng W, Zhang M, Organization and dynamics of PDZ-domain-related supramodules in the postsynaptic density. Nature Rev Neurosci 2009; 10: 87-99
Die Abbildung zeigt die Komplexität molekularer prä / postsynaptischer Verbindungen und Vernetzungen.
Präsynaptisch gibt
es neben der Exozytose des Transmitters auch calciumabhängige Kinasen.
Auf der postsynaptischen
Seite finden sich außer
Rezeptormolekülen und Ionenkanälen Adapter- und Signalproteine. Mehr als tausend
verschiedene Proteine können hier interagieren
und ihre Muster je nach Aktivierungsgrad der postsynaptischen Membran verändern.
Die beiden Membranen sind über Adhäsionsmoeküle (Cadherin, Neuroligin,
Neurexin, Ephrin, Ephrinrezeptor) miteinander verbunden. Diese
Verbindungen werden beständig auf- und abgebaut, je nach
Benutzungsintensität der Synapse (vgl. synaptische Plastizität).
AKAP, Adenylate-kinase anchoring protein, hilft bei der Anordnung von Signalproteinen
 BAR, hochkonservierte Proteindomäne
CaCh, Ca++-Kanal
CaMKII, calcium/calmodulin-dependent protein kinase II; an Lern- und Gedächtnisprozessen beteiligte serin / threoninspezifische Kinase
CASK, ein Membranprotein (calcium / calmodulin dependent serin protein kinase) CRIPT, cysteine-rich PDZ-binding protein; interagiert mit Synapsenproteine
BAR, hochkonservierte Proteindomäne
CaCh, Ca++-Kanal
CaMKII, calcium/calmodulin-dependent protein kinase II; an Lern- und Gedächtnisprozessen beteiligte serin / threoninspezifische Kinase
CASK, ein Membranprotein (calcium / calmodulin dependent serin protein kinase) CRIPT, cysteine-rich PDZ-binding protein; interagiert mit Synapsenproteinen
EphR, Adrenalinrezeptor
GKAP, guanylate kinase-associated protein; an der Errichtung postsynaptischer Verbindungen beteiligt
GRASP, GRIP-associated protein
GRIP, glutamate receptor interacting protein, Adaptermolekül für zellulären Transport IP3R, Inositol-1,4,5-trisphosphat-Rezeptor
KCh, K+-Kanal
MAP1A, microtubule-associated protein 1A, wichtig für Neurogenese
L27, Protein-Bindedomäne
mGluR, metabotroper Glutamatrezeptor
nNOS, neuronale NO-Synthase
NMDAR, N-Methyl-D-Aspartat-Rezeptor (Glutamatrezeptor)
PDZ, PDZ-Bindemotiv, mit anderen Proteinen interagierender Proteinteil (Proteininteraktionsdomäne)
PICK1, protein interacting with PRKCA1, Adapterprotein
SER, glattes endoplasmastisches Retikulum
SH3, Interaktionen vermittelnde Proteindomäne
SPAR, spine-associated RAPGA
SV, synaptisches Vesikel
SYNGAP, synaptic Ras GTPase-activating protein, an synaptischer Plastizität beteiligt
TIAM1, T-cell lymphoma invasion and metastasis -inducing protein 1, verknüpft extrazelluläre Signale mit Aktivitäten im Zytoskelett
TRAP, C-terminal receptor-binding region
Solche Brückenbildungen beeinflussen sowohl die Ausbildung
(Synaptogenese) als auch die Funktion (Ansprechgeschwindigkeit) von Synapsen. Zu den organisierenden /
beeinflussenden Molekülen zählen auch Immunglobuline und Wachstumsfaktoren. Diese steuern Organisation, Entwicklung und Funktion von Synapsen in
komplexer Weise. Dazu kommen zusätzliche
Einflüsse auf synaptische Aktivitäten durch Gliazellen.
Postsynaptischer Apparat
Synapsen erscheinen an der postsynaptischen Membran wegen ihres Reichtums an verschiedenen Proteinen (aktive Zone s. oben) elektronenmikroskopisch dicht (postsynaptic density PSD). Die PSDs verschiedener Synapsentypen beinhalten unterschiedliche Proteine (hunderte davon sind bekannt) und enthalten
auch Enzyme (Kinasen, Phosphatasen). Sie unterstützen die Asymmetrie
der Synapsenwirkung ("Einbahnsystem"). Im ZNS liegen mehr als 90% aller
Synapsen auf Dornenfortsätzen (dendritic spines),
die ankommende Information vor allem lokal wirksam machen.
Die folgende Abbildung zeigt die postsynaptischen Wirkungsmechanismen, welche Neurotransmitter am postsynaptischen Neuron auslösen können:
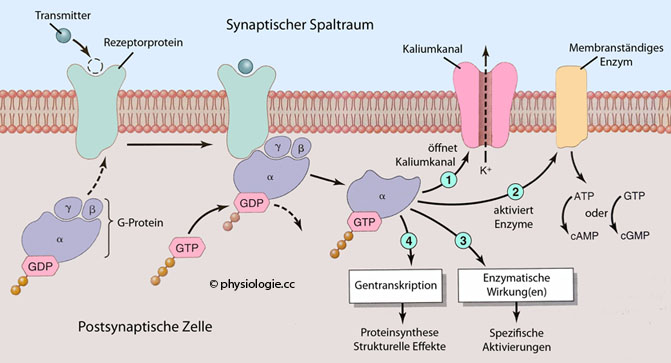
Abbildung: Postsynaptische Reaktionen auf die Bindung eines Neurotransmitters
Nach einer Vorlage bei Guyton and Hall, Textbook of Medical Physiology, 15th ed. Elsevier 2026
Koppelt ein Transmitter an seinen Rezeptor, ändert der Rezeptor seine Gestalt und aktiviert seine - an der Innenseite der Membran gelegene - Bindungsstelle
für den G-Protein-Komplex. Dieser koppelt daraufhin an den Rezeptor,
die
α-Untereinheit gibt (im Austausch gegen GTP) GDP ab, löst sich dabei
vom Komplex (d.h. von den zurückbleibenden Untereinheiten β und γ) ab,
kann sich frei im Zytosol bewegen und eine oder mehrere der folgenden
Wirkungen ausüben:
1. Öffnen von Ionenkanälen (in der Abbildung ein Kaliumkanal) und damit entsprechende Ionenströme durch die Membran
2.
Bildung von cAMP oder cGMP durch Enzymaktivierung. Dies kann
langwirkende Änderungen bewirken, z.B. der Erregbarkeit des Neurons
3. Aktivierung intrazellulärer Enzyme und damit entsprechender Stoffkonzentrationen in der Zelle
4.
Transkription von Genen mit entsprechender Proteinsynthese. Das kann
die Struktur und/oder den Stoffwechsel der Zelle beeinflussen und sich
u.a.auf das Langzeitgedächtnis auswirken
Nach seiner Freisetzung diffundiert der Transmitter über
den synaptischen Spalt zur subsynaptischen Membran (synaptische Wirkung), zurück zum präsynaptischen Apparat (reuptake), oder in den
umgebenden Extrazellulärraum, von dort in Liquor, Lymphsystem und Blutbahn (und werden in Blutproben nachweisbar). Er wird sowohl lokal als auch systemisch rasch wieder abgebaut (kurze Halbwertszeiten).
Neurotransmitter treffen postsynaptisch auf mehrere Rezeptor-Subtypen:
Für
Glutamat (ionotrop) NMDA, AMPA, Kainat, (metabotrop) mGluR
1 bis mGluR
6
Für
GABA GABA
A und GABA
B
Für
Acetylcholin muskarinerge (M
1 bis M
5) und nikotinerge (Muskel, neuronal: α-Bungarotoxin-insensitiv)
Für
Dopamin D
1 bis D
5
Für
Adrenalin / Noradrenalin α
1A bis α
1C, α
2A bis α
2D, ß
1 bis ß
3
Für
Serotonin 5-HT
1A bis 5-HT
1F, 5-HT
2A bis 5-HT
2C, 5-HT
3 bis 5-HT
7
Für
Histamin H
1 bis H
4
Für
Opioide µ
1 bis µ
3, δ
1, δ
2, κ
1 bis κ
3
Für
Endocannabinoide CB1 (Gehirn) und CB2 (Körperperipherie)
Zellen können so auf ein und denselben Signalstoff auf verschiedene Art
und Weise reagieren, die Antworten können sogar entgegengesetzt sein
(z.B. führt Reizung von D1-Rezeptoren zu erhöhter cAMP-Bildung, eine von D2-Rezeptoren
hemmt die cAMP-Synthese).
Genaue Kenntnis der Rezeptorverteilungen hat
große pharmakologische Bedeutung, da spezifisches Ansprechen bestimmter
Rezeptor-Subtypen wesentlich verfeinerte therapeutische Effekte
ermöglicht.
Der Transmitter kann postsynaptisch abgebaut
oder präsynaptisch wiederaufgenommen werden: Die Zellmembran unterliegt einem steten Recycling. Drei Mechanismen ermöglichen die rasche Beendigung der synaptischen Signalübertragung:
Diffusion und damit das Absinken der Transmitterkonzentration
Enzymatischer
Abbau im Bereich des synaptischen Spalts
Na
+-Gradient-abhängige
Aufnahme in den präsynaptischen Neuritenfortsatz
(reuptake, recycling) bzw. in benachbarte Gliazellen
(
Abbildung unten).
Praktisch alle Stufen des synaptischen Wirkmechanismus können chemisch beeinflusst werden (Neuropharmaka, Neurotoxine):
Aufnahmesysteme der Zellmembran (Aufnahme von Transmittervorstufen, Wiederaufnahme fertigen Transmitters)
Natriumkanäle (die Erregbarkeit der Nervenzelle kann durch
Lokalanästhetika - oder, spezifischer auf Na
+-Kanäle, durch
Tetrodotoxin - unterbrochen werden)
Calciumkanäle (Angriffspunkt z.B. von
Tetanusroxin,
Botulinustoxin)
Synthese- und Abbauenzyme
posttranslationale Reifung, axonaler Transport
vesikuläre Speicherung
Rezeptoren (präsynaptisch, postsynaptisch, unterschiedliche Rezeptortypen)
Plastizität: Rezeptoren
verändern ihre Ansprechbarkeit gegenüber Signalstoffen - das gilt auch
für Neurotransmitter. Das beruht auf mehreren Mechanismen: Up- oder
Downregulation; Phosphorylierung / Dephosphorylierung; Verschiebung von
rezeptorbeladenen Membranabschnitten zwischen "außen" und "innen".
Die Antworten dieser Rezeptoren unterscheiden sich stark in ihrer Wirkungsdauer: Millisekunden (rasche Transmission: Aminosäuren, cholinerg-nikotinisch), Sekunden (langsame Transmission: Katecholamine, cholinerg-muskarinisch), Minuten bis Tage (Fazilitation, Depression, Modulation). Je
länger die biologische Halbwertszeit, desto deutlicher tritt die
endokrine Komponente eines Transmitters in Erscheinung (z.B.
Noradrenalin, Vasopressin).
Fazilitation bedeutet eine kurzfristige (10-100 Millisekunden) Erhöhung, Depression
eine Erniedrigung der synaptischen Wirkung nach hochfrequenter Reizung
des betreffenden Synapsensystems. Habituation ist eine allmähliche Abschwächung im Rahmen andauernder relativ schwacher (niedrigfrequenter) Reizung.
Diese Phänomene erklären sich durch
Verstärkung oder Abschwächung der Transmitterfreisetzung und der
Rezeptoransprechbarkeit mit jeder folgenden Erregung. Fazilitation und
Depression sind Mechanismen der synaptischen Plastizität: Der Abhängigkeit der Übertragungseffizienz in Abhängigkeit von der Vorgeschichte an der Synapse.
Augmentation ist eine Verstärkung der Synapsenwirkung über mehrere Sekunden, der Effekt einer posttetanischen Potenzierung kann bis zu mehrere Minuten nach der Reizung anhalten.
Zu posttetanischer Potenzierung und synaptischer Plastizität s. auch dort
Zu Neuromodulation s. unten
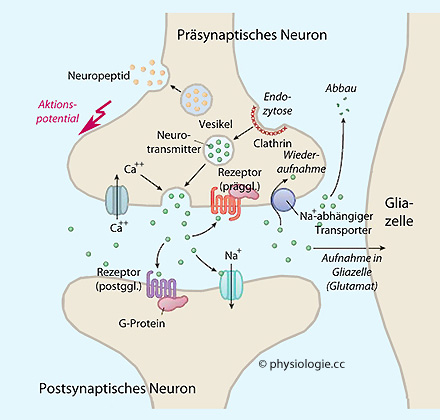
Abbildung: Freisetzung, Wirkung und Inaktivierung von Neurotransmittern
Nach einer Vorlage bei Hilal-Dandan / Brunton, Goodman
& Gilman's Manual of Pharmacology and Therapeutics, 2nd ed., McGraw
Hill Education 2014
Depolarisierung der präsynaptischen Zelle (Eintreffen eines Aktionspotentials) führt zu Ca++-Einstrom
und dieser zu Exozytose des Transmitters. Dieser bindet postsynaptisch
an G-Protein-gekoppelte oder an Ionenkanal-Rezeptoren, was
entsprechende Effekte am postsynaptischen Neuron hervorruft.
Präsynaptisch bindet der Neurotransmitter an G-Protein-gekoppelte Rezeptoren, die dann die Transmitterfreisetzung modifizieren (hemmen oder fördern); oder an Na+-Symporter, die ihn wiederaufnehmen.
Gliazellen können Transmitter ebenfalls aufnehmen, falls sie entsprechende Transporter exprimieren
Neurotransmitter
werden hingegen nicht nur aus Vorstufen im Soma gebildet (z.B. Dopamin
aus Tyrosin) und ebenfalls axonal transportiert, sondern auch nach
synaptischer Freisetzung peripher wiederaufgenommen (reuptake),
oder hier aus Vorstufen (z.B. Glutamin für Glutamat, Cholin für Acetylcholin) zusammengesetzt. Weiters können extrazellulär
auftauchende Neurotransmitter von Gliazellen aufgenommen werden
( Abbildung).
Präsynaptische vs. postsynaptische Rezeptoren
Neurotransmitter können an präsynaptischen (Auto-) oder postsynaptischen Rezeptoren angreifen.

Abbildung
: Wirkung eines Neurotransmitters an präsynaptischen Rezeptoren
Nach einer Vorlage in Kandel / Koester / Mack / Siegelbaum (eds), Principles of Neural Sciences, 6th ed. 2021 (McGraw Hill)
Präsynaptische metabotrope Rezeptoren bilden second messengers. Diese können die Effizienz der Transmitterfreisetzung auf verschiedenen Wegen beeinflussen: Durch direkte Beeinflussung von Ca++-Kanälen oder indirekt durch Wirkung auf K+-Kanäle, was das Membranpotential und damit den Ca++-Einstrom
ändert - das hat Auswirkung auf die Länge des Aktionspotentials
(rechts). Dauert das präsynaptische Aktionspotential länger, erhöht
sich postsynaptisch das EPSP
Ionotrope Rezeptoren - z.B. nikotinartige cholinerge Rezeptoren - beeinflussen über die Permeabilität von Ionen direkt das Membranpotential und finden sich üblicherweise in der postsynaptischen Membran. Sie wirken
rasch (Millisekunden) - begrenzt auf ihr unmittelbares Umfeld - entweder de- (EPSP: exzitatorisch) oder hyperpolarisierend (IPSP: inhibitorisch).
Metabotrope Rezeptoren - etwa 80% aller Neurotransmitter und Neurohormone - wirken über G-Proteine auf intrazelluläre second-messenger-Mechanismen und funktionieren etwas
langsamer (Sekunden). Sie bilden Botenstoffe, die durch
Diffusion auch in einiger Entfernung vom aktivierten Rezeptor und
unterschiedliche Ionenkanäle beeinflussen können. Oft wirken sie auf die präsynaptische Neuronenendigung und modulieren dort die Freisetzung des Neurotransmitters. So kann z.B. der präsynaptische Einstrom von Ca++
direkt oder indirekt beeinflusst und damit die synaptische Intensität
gesteuert werden.

Abbildung: Wirkung von Noradrenalin auf präsynaptische Calciumkanäle
Nach einer Vorlage in Liqun Luo, Principles of Neurobiology, 2nd ed. CRC Press 2021
Von
einer postganglionär-sympathischen Nervenzelle freigesetztes
Noradrenalin bindet an präsynaptische adrenerge α-Rezeptoren derselben
Zelle (links). Der dadurch freigewordene βγ-Komplex des G-Proteins
diffundiert zu Calciumkanälen in der benachbartern Membran und senkt
deren Öffnungswahrscheinlichkeit - beim Eintreffen von
Aktionspotentialen strömen weniger Calciumionen in die Zellle ein.
Der sinkende Ca++-Spiegel in der präsynaptischen Endigung
senkt die Häufigkeit der Vesikel-Exozytose und damit die Freisetzung
von Noradrenalin. Die Synapse hat ihre Übertragungseffizienz gesenkt
(negative Rückkopplung)
Die Abbildung
zeigt das Beispiel der Selbsthemmung adrenerger Synapsen durch Senkung
des Calciumeinstroms durch freigesetztes Noradrenalin. So können
präsynaptische Nervenendigungen bei anhaltender Aktivität (hoher
Aktionspotentialfrequenz) durch negatives Feedback die eigene Freisetzung ihres Neurotransmitters reduzieren - eine synaptische Kurzzeit-Plastizität.
Präsynaptische Axonendigungen können auch metabotrope Rezeptoren (also GPCRs) für Transmitter enthalten, die von anderen Nervenendigungen freigesetzt werden (Abbildung
). So sind sowohl Bahnungs- als auch Hemmeffekte möglich, welche die Synapsenaktivität beeinflussen - je nach Art des Neurotransmitters, der involvierten Rezeptoren, Signalwege und Effektoren (z.B. Enzyme) in der Zielzelle.

Abbildung
: Präsynaptische Bahnung und Hemmung
Nach einer Vorlage in Liqun Luo, Principles of Neurobiology, 2nd ed. CRC Press 2021
Links: Metabotrope
Beeinflussung der präsynaptischen Membran kann hier eine Bahnung
hervorrufen. In diesem Beispiel senkt Serotonin über einen
Serotonin-GPCR an der Endigung eines präsynaptischen Neurons die
Öffnungswahrscheinlichkeit von Kaliumkanälen und senkt dadurch das
Membranpotential (wirkt also depolarisierend).
Rechts: Metabotrope Beeinflussung der präsynaptischen Membran kann auch inhibierend wirken. In diesem Beispiel wirkt GABA über GABAA-Rezeptoren
die Öffnungswahrscheinlichkeit von Chloridkanälen in der Membran des
präsynaptischen Neurons (Mitte) und senkt damit die
Depolarisierungsgröße (den Effekt) bei Eintreffen eines
Aktionspotentials. Wirkung auf GABAB-Rezeptoren
kann die Öffnung von Kaliumkanälen wahrscheinlicher oder diejenige von
Calciumkanälen seltener machen; beides hat einen hyperpolarisierenden
(inhibitorischen) Effekt
Werden Kaliumkanäle geschlossen, nimmt das Membranpotential (das durch K+-Ausstrom
aufrechterhalten wird) ab und die Membran wird depolarisiert; das
erleichtert die Aktivierung spannungsabhängiger Calciumkanäle und regt
die Freisetzung des Neurotransmitters an. Man spricht von
präsynaptischen Effekten (presynaptic facilitation / inhibition).
An
der postsynaptischen Membran können metabotrope Rezeptoren die Amplitude der hervorgerufenen Potentialänderungen (EPSP, IPSP) über Wirkung auf ionotrope Rezeptoren variieren ( Abbildung
). Dadurch steuern sie - an Dendriten,
Soma, oder Axon - mehrere Größen (Membranwiderstand, Längskonstante, Ruhepotential, Schwellenpotential, Aktionspotentialdauer).
Neuromodulation
Die
Informationsübertragung an Synapsen wird oft durch zusätzlich in den
Synapsenraum abgegebene Stoffe beeinflusst, die dort an der prä- und
postsynaptischen Membran und an subzellulären Kompartimenten ihrer
Zielneurone wirken und eine Fülle von Effekten erzielen können. Solche
Stoffe werden als Neuromodulatoren bezeichnet. Dabei handelt es sich um

Transmitter, die in der
Peripherie des - den Neuromodulator
produzierenden - Axons synthetisiert oder wiederverwendet und in
Vesikeln gespeichert werden, wie Monoamine (
Noradrenalin,
Dopamin,
Serotonin,
Histamin) und
Acetylcholin; oder
Neuropeptide (mehr als einhundert sind bekannt) mit 2 bis 50 Aminosäuren, z.B.
CCK,
α-MSH,
NPY,
AgRP). Diese werden im
Soma der Nervenzelle
synthetisiert (nahe an Zellkern und endoplasmatischem Retikulum) und anschließend zum synaptischen Terminal
transportiert.
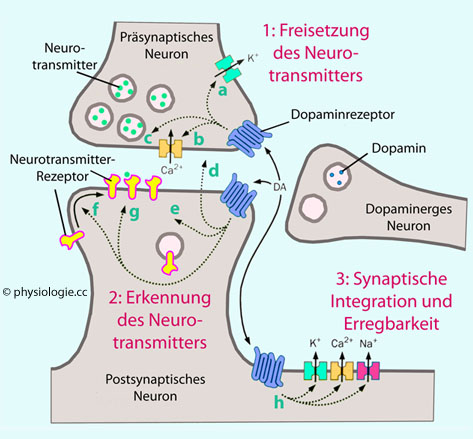
Abbildung: Wirkung von Neuromodulatoren auf Zielneurone
Nach einer Vorlage in Liqun Luo, Principles of Neurobiology, 2nd ed. CRC Press 2021
Beispiel Dopamin (DA), das von einem dopaminergen Nervenende (rechts) freigesetzt wird. Es beeinflusst als Neuromodulator..
(1) ..die präsynaptische Freisetzung
eines Neurotransmitters durch Wirkung auf Kaliumkanäle (a),
Calciumkanäle (b), Freisetzung des Inhals von Vesikeln (c) oder die
postsynaptische Produktion eines retrograd wirkenden Botenstoffs (d);
(2) die postsynaptische Erkennung des Neurotransmitters durch Wirkung auf seine Rezeptoren: Einlagerung in die (e), Bewegung zur subsynaptische(n) Membran (f), Veränderung ihrer Leitfähigkeit (g);
(3) die synaptische Integration und Erregbarkeit des postsynaptischen Neurons durch Wirkung auf spannungsgesteuerte Ionenkanäle (h)
(Neuro-) Modulation ist die Abänderung neuronaler Aktivität durch Wirkung eines Neuromodulators, meist über metabotrope - G-Protein-gekoppelte und daher langsamer wirkende - Rezeptoren (GPCRs) an ihren Zielzellen. Diese können sowohl auf der Membran präsynaptischer als auch postsynaptischer Neuronen liegen. So können
metabotrope Rezeptoren durch
Modulation ionotroper Rezeptoren in der postsynaptischen Membran die Öffnungswahrscheinlichkeit ionotroper Rezeptoren und damit die Amplitude postsynaptischer Potentiale verändern. Allgemein kann man Produkte von Nervenzellen, welche die Aktivität anderer Nervenzellen beeinflussen, als natürliche neuroaktive Substanzen (NAS, natural neuroactive substances) bezeichnen. Neuromodulatoren können auch gemeinsam mit "klassischen" Transmittern freigesetzt werden (Cotransmission).
Neuromodulation beeinflusst die
Freisetzung oder Wirkung von Neurotransmittern
(die im Millisekundenbereich
wirken) auf einer langsameren Zeitskala (Sekunden bis Tage). So werden
nachhaltige (bis
zu mehrere Tage andauernde) Effekte bewirkt: Ablesung
von Genen (Transkription) und Proteinsynthese (Translation) und
Neubildung von Ionenkanälen, Einlagerung in die synaptische Membran,
Intensivierung des
Ansprechverhaltens etc ( Abbildung
). Neuromodulation wirkt längerfristig und ist auch bei Gedächtnisprozessen involviert.

Abbildung
: Neuromodulation sympathischer / parasympathischer Signalübertragung
Nach einer Vorlage in Herring / Paterson, Levick's Introduction to Cardiovascular Physiology, 6th ed. 2018
Die Freisetzung von Noradrenalin (NA) und Acetylcholin (ACh) aus postganglionären Neuronen wird angeregt durch Calciumionen,
die - insbesondere bei Eintreffen von Aktionspotentialen - durch
spannungsgesteuerte Ionenkanäle (graue Boxen) in die Zelle
gelangen.
Neuromodulatoren
können aus Blutgefäßen stammen, z.B. Angiotensin (Ang II) oder C-Typ
natriuretisches Peptid (CNP); aus Myozyten, z.B. B-Typ
natriuretisches Peptid (BNP); oder aus Neuronen, z.B. Neuropeptid Y
(NPY), Galanin (Gal), vasoaktives intestinales Peptid (VIP), oder
(intrazellulär) neuronale NO-Synthase (nNOS) bzw. das Aktivatorprotein
CAPON (carboxy-terminal PDZ ligand of nNOS).
Grüne
Pfeile symbolisieren anregende, rote Pfeile hemmende modulatorische Wirkpfade
Neuromodulation erfolgt nicht nur im Zentralnervensystem, sondern auch in der Peripherie des autonomen Systems (Abbildung
). Dabei können die Neuromodulatoren auf die Freisetzung oder Aufnahme von
Transmittern wirken, autokrin oder parakrin, anregend oder
(auto-)inhibitorisch.
Neuromodulatorische Systeme und Mechanismen sind erst ansatzweise
verstanden und Gegenstand intensiver Forschung. Ihre Bedeutung spiegelt
sich in der Tatsache wider, dass die meisten Medikamente, die zur
Behandlung psychiatrischer Störungen Verwendung finden, mit der
Funktion von Neuromodulatoren interferieren.
Postsynaptische Potentiale, Summation, Genexpression
Zu postsynaptschen Potentialen s. auch dort
Die Ankunft von Aktionspotentialen an
axonalen Enden löst Vorgänge aus, die auf das folgende Neuron
exzitatorisch oder inhibitorisch
wirken. An der postsynaptischen Membran
verändert sich als Folge der Durchtritt von Ionen und damit das
Membranpotential. Die Wirkung kann eine depolarisierende oder
hyperpolarisierende , erregende oder hemmende sein.
Kleine Nichtpeptide als Neurotransmitter werden großteils in
der Nähe des Synapse synthetisiert, vesikulär gespeichert und bei präsynaptischer
Erregung - unter Vermittlung von Ca++-Ionen - in den synaptischen Spaltraum exozytotisch freigesetzt. Dabei entsteht ein docking complex
aus verschiedenen Proteinen - Neurexin, Syntaxin, Rab3, Synaptobrevin,
Synaptotagmin (s. unten) und ein Transmitter-Transporter ( Abbildung).

Abbildung: Erregende und hemmende Neurotransmission
Nach einer Vorlage bei Hilal-Dandan / Brunton, Goodman
& Gilman's Manual of Pharmacology and Therapeutics, 2nd ed., McGraw
Hill Education 2014
Links: Ein Aktionspotential trifft über eine präsynaptische Afferenz an der Synapse ein. Dies führt
zur Freisetzung eines Transmitters, der exzitatorisch (oben) oder inhibitorisch (unten)
wirkt: Das eintreffende präsynaptische Aktionspotential (1. Ap) bewirkt
bei einer exzitatorischen Synapse postsynaptisch ein EPSP und Bahnung
(bei Überschwelligkeit ein Aktionspotential), an einer inhibitoríschen
Synapse ein IPSP.
Mitte: Details der involvierten Ionenkanäle. Oben der Anlagerungskomplex, der die Exozytose des
Transmitters ermöglicht, unten ein postsynaptischer
spannungssensitiver Natriumkanal. Rechts: Benennung der verwendeten graphischen Symbole
Peptide werden hingegen im Zellsoma gebildet (
Translation) und per
axonalem Transport in die Peripherie gebracht.
Einzelne postsynaptische Potentiale haben eine kleine Amplitude (0,01-1 mV) und bewirken dementsprechend nicht viel. Summation mehrerer postsynaptischer Potentiale hingegen wirkt sich stärker aus; sie kann die Zelle bis über ihr Schwellenpotenial
hinaus depolarisieren und so ein Aktionspotential hervorrufen.
 Wenn z.B. an einem Neuron die Aktivierung einer exzitatorische Synapse
das Membranpotential um 1 mV reduziert und das Schwellenpotential 15 mV
vom Ruhepotential entfernt liegt, bedarf es an dieser Nervenzelle der
Wirkung von mindestens 15 EPSPs, um ein Aktionspotential auszulösen.
Wenn z.B. an einem Neuron die Aktivierung einer exzitatorische Synapse
das Membranpotential um 1 mV reduziert und das Schwellenpotential 15 mV
vom Ruhepotential entfernt liegt, bedarf es an dieser Nervenzelle der
Wirkung von mindestens 15 EPSPs, um ein Aktionspotential auszulösen.

Abbildung: Räumliche und zeitliche Bahnung
Nach einer Vorlage in Boron / Boulpaep: Concise Medical Physiology, Elsevier 2021
Eintreffende einzelne Aktionspotentiale
bewirken am betreffenden Dendrit und am Soma des Neurons exzitatorische
postsynaptische Potentiale (PSPs), die unterschwellig bleiben (A).
Gelangen Aktionspotentiale über mehrere Eingänge an die Nervenzelle,
erhöht deren Einfluss das PSP (räumliche Summation), sodass dieses
überschwellig wird und am Axonhügel Aktionspotentiale auslöst, die über
das Axon weitergeleitet werden (B, C).
Knapp aufeinander folgende Erregungen können das PSP ebenfalls überschwellig werden lassen (C)
Treffen
präsynaptisch mehrere Erregungen knapp hintereinander ein, sind die auf
den ersten Impuls folgenden Effekte (Transmitterfreisetzung) verstärkt:
Die terminale [Ca++] (im präsynaptischen
Zytoplasma) ist nach der ersten Erregung noch erhöht ("Restcalcium"),
und es wird beim nächsten Engagement der spannungsgesteuerten Calc
iumkanäle der präsynaptischen Membran mehr Neurotransmitter
freigesetzt. Das verstärkt postsynaptisch den EPSP-Effekt.
Dieses
Phänomen wird als eine der Grundlagen für synaptische Plastizität und Kurzzeitgedächtnis gesehen.
Ein zweites kurz nach einem vorangehenden EPSP ist größer als das erste, weil die präsynaptisch- zytoplasmatische Ca++-Konzentration noch erhöht ist
|
Das Gegenstück dazu ist synaptische Depression oder Habituation (synaptic depression).
Dabei handelt es sich um eine vorübergehende Abnahme der Effizienz, mit
der bei Erregung der postsynaptischen Zelle Transmitter freigesetzt
wird. Sie wird vor allem an inhibitorischen Synapsen beobachtet und
erklärt sich mit einer abnehmenden Transmitterbeladung der Vesikel,
üblicherweise infolge starker vorangegangener Entleerung.
Durch
Hyperpolarisation wird eine Nervenzelle (oder glatte Muskelzelle) gehemmt und bildet weniger
oder
keine Aktionspotentiale. Inhibierende Synapsen setzen einen Transmitter
frei, der postsynaptisch zu jeweils einem inhibitorischen
postsynaptischen Potential (IPSP)
führt. Diese Hyperpolarisierung erfolgt meist durch erhöhte Durchlässigkeit für Chloridionen (Einstrom von Anionen), oder auch durch einen Anstieg der Kaliumdurchlässigkeit der Membran (Ausstrom von Kationen). Jedes IPSP
trägt dazu bei, das Membranpotential der Nervenzelle zu stabilisieren
bzw. zu erhöhen, und sie so gegenüber anregenden Reizen weniger
empfänglich zu machen.

Die meisten Synapsen finden sich an Dendriten, mit einem Axon-Endfortsatz (axonalem Terminal) als präsynaptischem, und einem Dornenfortsatz (dendritic spine) als postsynaptischem Anteil.

Abbildung: Abschwächung postsynaptischer Potentiale in Dendriten
Nach einer Vorlage in Boron / Boulpaep: Concise Medical Physiology, Elsevier 2021
Ein Neuron (oben) sendet Aktionspotentiale, die an Dendriten zweier
anderer Neurone eintreffen (unten). Die eintreffenden exzitatorischen
postsynaptischen Potentiale (EPSPs) haben identische Größe (obere
Registrierungen).
Das linke Neuron hat dünne Dendriten mit einer geringen Längskonstante
(λ), das rechte dickere mit einem höheren λ-Wert (entsprechende Länge
mit "l" angezeigt). Daher ist der Effekt am Axonhügel des linken
Neurons schwach (und unterschwellig), an dem des rechten Neurons
überschwellig (ein Aktionspotential wird ausgelöst - untere
Registrierungen)
V
m, Membranpotential; Abszisse: Zeit
Dendriten unterscheiden sich in Länge und Durchmesser, und damit in der Längskonstante
(λ): Dicke Dendriten leiten Strom besser (zum Soma) als dünne
(
Abbildung). Beispielsweise weist ein Dendrit mit 0,2 µm
Durchmesser einen λ-Wert von 0,35 mm, ein Dendrit mit 10 µm Durchmesser
(ceteris paribus) einen λ-Wert von 2,5 mm
auf - etwa der 7-fache Wert. Damit werden EPSPs dicker Dendriten auch
stärker zur Depolarisierung des Axonhügels beitragen als solche an
dünnen.
Dazu kommt, dass Dendriten eher stetige Potentiale besser leiten als
sich rasch ändernde - sie wirken als Tiefpassfilter: Rasch variierende
Potentialsignale haben einen verhältnismäßig geringeren Effekt.
Axonhügel und Auslösung eines Aktionspotentials: Der Betrag des Schwellenpotentials (threshold potential)
ist am Axonhügel eines Neurons - der Stelle der Nervenzelle, an der
meist Aktionspotentiale entstehen (Triggerzone) - im Allgemeinen um
10-15 mV geringer als der des Ruhepotentials. (Die Membran des Soma und
der Dendriten ist weniger erregbar - hier liegt das Schwellenpotential
bis zu 30 mV vom Ruhepotential entfernt.) An den Axonhügel schließt sich eine 15 bis 50 µm lange myelinfreie Zone an - der Beginn des Axons ist unmyelinisiert. Durch dieses initiale Segment
fließen Reizströme, die eine überschwellige Depolarisation triggern; es
dient als "Brennpunkt" für die Entstehung von Aktionspotentialen.
Das Aktionspotential läuft von hier orthodrom über das Axon und dessen Verzweigungen bis zu den Endstücken (axon terminals) mit den präsynaptischen Apparaten; sowie - abgeschwächt -
antidrom
über das Soma und die Dendriten der Nervenzelle. Die orthodrome Leitung
dient der Signalleitung, die Funktion der antidromen ist nicht ganz
klar (Löschen laufender Verrechnungsprozesse, Modifikation von
Membraneigenschaften, Einfluss auf synaptische Plastizität..?).
Ändert sich das
Ruhepotential, dann verschiebt sich auch das Schwellenpotential -
abhängig vom Spannungsbetrag und von der Geschwindigkeit der
Potentialänderung. Depolarisiert man eine Membran langsam, gleitet das
Schwellenpotential sozusagen davon ("Akkommodation"); die
Depolarisierung muss rasch genug erfolgen, man spricht vom einem Steilheitsbedarf. Bei ausreichender (und ausreichend rascher) Depolarisation über das Schwellenpotential hinaus
wird das postsynaptische Neuron erregt und bildet ein Aktionspotential.
Postsynaptische Depolarisierung kann die Expression von Genen anregen.
Neben kurzfristigen Potentialänderungen an der postsynaptischen Membran
(über ionotrope Rezeptoren für Millisekunden, über metabotrope für
Millisekunden bis Sekunden) können Neurotransmitter auch
bewirken, dass im postsynaptischen Apparat neue Gene exprimiert werden (Abbildung). Diese Wirkung hält wesentlich länger an (vgl. auch dort).

Abbildung: Signalwege von der Synapse zum Zellkern
Nach einer Vorlage in Liqun Luo, Principles of Neurobiology, 2nd ed. CRC Press 2021
Die dargestellten Signalwege führen zur Phosphorylierung von CREB.
Bindet Glutamat an NMDA-Rezeptoren der postsynaptischen Membran (auf
Dornenfortsätzen der Dendriten), strömen Calciumionen in die Zelle ein.
Die resultierende Depolarisierung öffnet weiters spannungsabhängige
Calciumkanäle des Dendriten, die intrazelluläre [Ca++]
steigt an. Auch aus dem endoplasmatischen Retikulum treten Calciumionen
durch Ryanodinrezeptoren (calciumsensitive Calciumkanäle) in das
Zytoplasma ein.
An Calmodulin gebundenes Ca++ aktiviert CaM-Kinasen, Rsk
(Ras-MAP-Kinase) und Proteinkinase A (über cAMP). Diese Faktoren führen
zu Phosphorylierung von CREB, welches anschließend Zielgene mit CREs in
entsprechenden Promotoren anregt
Die verschiedenen in der
Abbildung
gezeigten Signalwege von der Synapse bis zur Freigabe der Transkription
diverser Gene im Zellkern des Zielneurons involvieren gemeinsam erhöhte
Calciumspiegel, haben aber unterschiedliche funktionelle Eigenschaften.
So funktioniert der Calmodulin-Kinase-Weg rasch (innerhalb von Minuten
nach der Depolarisierung kommt es zur Phosphorylierung von CREB),
während sich die Wirkung des MAP-Kinase-Weges über Stunden hinzieht.
Außer CREB können weiters andere calciumsemsitive
Transkriptionsfaktoren an unerschiedliche Promotorregionen binden.
So kann die (präsynaptische) neuronale Aktivität über verschiedene Signalwege (postsynaptisch) auf den Zellkern des Zielneurons wirken und nicht nur direkt dessen
Transkriptionsmuster beeinflussen, sondern auch epigenetische Modifikationen
vornehmen - über Enzyme, welche die Methylierung der DNA und
posttranslationelle Modifikation der Histone (Methylierung /
Demethylierung, Acetylierung / Deacetylierung) regeln und so - über die
Zugänglichkeit zu Gensequenzen - das Muster der Genexpressionen
beeinflussen. Das hat längerfristige Folgen für die Funktion der
betroffenen Nervenzellen.
Entfernung des Transmitters aus dem synaptischen Spaltraum
Wie geht es mit dem Transmitter weiter? Transmittermoleküle werden nach ihrer Freisetzung vom präsynaptischen
Apparat
teils an Rezeptoren gebunden,
dann endozytiert und abgebaut;
teils werden sie präsynaptisch gebunden (können dort auch negativ
rückkoppelnd wirken) und
wieder aufgenommen (recycling);
teils
diffundieren sie in den Extrazellulärraum weiter und werden z.T. dort
enzymatisch inaktiviert,
oder mit dem Kreislauf weitertransportiert und
können so - auf größere Distanz - auch neuroendokrin aktiv werden.
I
n den synaptischen Spaltraum freigesetzte Transmitter müssen
rasch wieder entfernt werden, um die Signalübertragung nicht zu
blockieren (längere Bindung an Rezeptoren führt rasch zu deren
Herunterregulierung) und postsynaptische Refrakterität zu vermeiden.
Das erfolgt durch Diffusion in die Umgebung (im Gehirn wegen der engen
Nachbarschaft zu anderen Synapsen kein besonders effizienter
Mechanismus), enzymatischen Abbau, sowie Wiederaufnahme (reuptake) in den präsynaptischen Apparat - der wichtigste Mechanismus für die rasche Entfernung kleiner Neurotransmitter.
Diese Transporter für Aminosäuren und biogene Amine weisen spezifische
Funktion, Lokalisation und Pharmakologie auf. Sie nutzen Symport mit Na+ (neurotransmitter sodium transporters), manche zusätzlich K+-, Cl-- oder H+-Transport. Beispielsweise wird ein Glutamatanion zusammen mit 3 Na+ und 1 H+ gegen Austausch mit 1 K+ befördert (was den Import von zwei positiven Ladungen bedeutet - das Membranpotential unterstützt diesen Austausch).
Kokain
blockiert die präsynaptische Wiederaufnahme von Serotonin, Noradrenalin
und Dopamin und verlängert ihre Wirkung; das Antidepressivum
Fluoxetin inhibiert selektiv die Aufnahme von Serotonin.
Die synaptische Konzentration von Glutamat wird auf mehrfache Weise reguliert: Es wird von Gliazellen
aufgenommen, zu Glutamin umgewandelt und Nervenzellen in dieser Form
wieder zur Verfügung gestellt (diese bilden daraus neues Glutamat). Acetylcholin
wird im synaptischen Raum von Acetylcholinesterase aufgespalten; das
resultierende Cholin kann präsynaptisch aufgenommen und wiederverwertet
werden. Aminotransmitter werden teils wiederverwendet, teils durch die Enzyme MAO und COMT abgebaut.
 Glutamat ist der führende Transmitter des Gehirns: Exzitatorische Synapsen im Zentralnervensystem funktionieren zum
größten Teil glutamaterg; Glutamat ist der Neurotransmitter jeder zweiten Synapse im Zentralnervensystem.
Glutamat ist der führende Transmitter des Gehirns: Exzitatorische Synapsen im Zentralnervensystem funktionieren zum
größten Teil glutamaterg; Glutamat ist der Neurotransmitter jeder zweiten Synapse im Zentralnervensystem.
Glutamat ist eine nichtessentielle Aminosäure, welche nicht direkt aus dem Kreislauf in Nervenzellen übertreten kann (Blut-Hirn-Schranke). Neuronen
synthetisieren Glutamat aus lokal verfügbaren Vorläuferverbindungen -
vor allem aus Glutamin. Dieses nehmen präsynaptische Nervenendigungen
aus dem Extrazellulärraum über einen System A-Transporter (SAT2) auf -
SAT2 beeinflusst wesentlich die Balance zwischen verfügbarem Glutamin
und Glutamat.
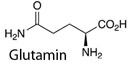 Glutamat ist nicht nur Transmitter, sondern (zusammen mit Asparagin) auch wichtiger Baustein zerebraler Proteine. Glutamat kann aus Glucose gewonnen werden (Citratzyklus: Transaminierung von 2-Oxoglutarat), und über die Synthese von Glutamin entfernen Astrozyten (mittels Glutaminsynthetase aus Glutamat + NH3) Ammoniak aus dem Nervengewebe.
Glutamat ist nicht nur Transmitter, sondern (zusammen mit Asparagin) auch wichtiger Baustein zerebraler Proteine. Glutamat kann aus Glucose gewonnen werden (Citratzyklus: Transaminierung von 2-Oxoglutarat), und über die Synthese von Glutamin entfernen Astrozyten (mittels Glutaminsynthetase aus Glutamat + NH3) Ammoniak aus dem Nervengewebe.
Glutamat wirkt auf verschiedene (rasche und langsame) postsynaptische Mechanismen (Divergenz),
und zwar über vier Klassen von Rezeptoren (R): Eine metabotrope (mGluR) und
drei ionotrope (NMDA-, AMPA- und Kainat-Rezeptoren).
Die Rezeptoren sind (wie auch
andere Rezeptortypen) mittels Adapterproteinen wie PSD-95 mit dem Zytoskelett und miteinander verknüpft. Das erleichtert das Clustering - die Ausbildung von Rezeptorgruppen ("Metabolons") - in der Zellmembran.
Ionotrope
Glutamatrezeptoren, die nach Rezeptor-Agonisten - AMPA, NMDA und Kainat
- bezeichnet worden sind (diese Pharmaka wirken spezifisch - sie kommen im Körper nicht vor, die Rezeptoren wurden aber nach ihnen benannt), wirken exzitatorisch. Geöffnete ionotrope Glutamatrezeptoren erlauben die Passage von Na+ (in die Zelle) und K+ (aus der Zelle), der Gesamteffekt ist eine Depolarisierung; geöffnete NMDA-Rezeptoren sind auch für Ca++-Einstrom zugänglich ( Abbildung).
Das erinnert an die Wirkung nikotinischer Acetylcholinrezeptoren (nAChR). Beide gehören einer umfangreichen Rezeptorgruppe an (glutamaterge,
cholinerge, GABA-, Glycin-, ATP-Rezeptoren), deren Gene
unterschiedlich gespleißt werden - daraus ergibt sich eine Vielfalt spezialisierter Rezeptoren.
Die
meisten exzitatorischen Synapsen im Gehirn haben sowohl AMPA- als auch
NMDA-Rezeptoren; NMDA-produzierte Depolarisierungen (EPSCs, excitatory postsynaptic currents) sind schwächer,
dauern aber länger als durch AMPA-Rezeptoren ausgelöste. Kainat-Effekte
sind ebenfalls schwach und dauern auch kurz; AMPA-Rezeptoren sind daher
die primären Träger exzitatorischer Übertragung im Gehirn.

Abbildung: Ionotrope Glutamatrezeptoren (mit direktem Gating)
Nach einer Vorlage in Kandel / Koester / Mack / Siegelbaum (eds), Principles of Neural Sciences, 6th ed. 2021 (McGraw Hill)
Der Rezeptorkomplex ist tetramer,
seine 4 Untereinheiten haben je 3 transmembranale α-Helices und eine
Aminosäureschleife,
die in die Membranebene reicht und wesentlich zum Aufbau des zentralen
Ionenkanals beiträgt. Einige dieser ionotropen Rezeptoren verfügen über
mehr als nur eine Bindungsstelle (bis zu 4) für Glutamat, was die
Bindungskinetik komplizierter macht (es kann mehr als nur einen Öffnungszustand mit entsprechend definierter Leitfähigkeit geben).
Links:
Der AMPA / Kainat-Rezeptortyp bindet Glutamat (Glu) und gibt den Kanal für die Passage von Kationen (Na+, K+)
frei. Da der Natriumgradient dominiert, bewirkt die Kanalöffnung bei
einem Membranpotential in der Nähe des Ruhepotentials vor allem einen
Natriumeinstrom; damit steht der depolarisierende Effekt im Vordergrund.
Rechts: Der geöffnete NMDA-Rezeptor lässt Na+, K+ und Ca++ passieren. Er verfügt über Bindungsstellen nicht nur für Glutamat, sondern auch für Glycin (Gly), Zink, Magnesium, Phencyclidin (PCP). Jede dieser Substanzen beeinflusst den Rezeptor in spezifischer Weise
Ionotrope Glutamatrezeptoren
sind tetramer, sie bestehen aus vier Subabschnitten mit jeweils einer
membranständigen Helix, die um einen zentralen Kanal angeordnet sind.
Der AMPA-Rezeptor ist in 4, der Kainatrezeptor in 5 verschiedenen Genen
codiert. AMPA- und NMDA-Rezeptoren werden in der postsynaptischen
Membran durch Proteine organisiert, die Teil der postsynaptischen
Verdichtungszone (postsynaptic density) sind. Hier befinden sich u.a. TARP (transmembrane AMPA receptor regulatory proteins) und das postsynaptic density protein PSD-95 (95 kDa), die mit Glutamatrezeptoren interagieren.
Die Mehrzahl der glutamatergen Synapsen bilden EPSPs (exzitatorische postsynaptische Potentiale), die aus zwei Komponenten bestehen ( Abbildung). Diese unterscheiden sich in Ionenbeteiligung, Abhängigkeit
von der Membranspannung, Kinetik und pharmakologischem Profil. Auch
dienen sie im Gehirn unterschiedlichen Funktionen. Ionotrope
Glutamatrezeptoren sind aus einem "Bausatz" von 14 verschiedenen
Untereinheiten zusammengefügte Heterotetramere: Je vier davon sind
jeweils um einen zentralen Ionenkanal angeordnet.
Abbildung: Glutamat-getriggerte Ionenkanäle
Nach einer Vorlage in Boron / Boulpaep: Concise Medical Physiology, Elsevier 2021
An
den meisten glutamatergen Synapsen wirken zwei Rezeptortypen bei der
Bildung eines EPSP
zusammen: AMPA-Ionenkanäle, die eine rasche, und NMDA-Kanäle, die eine
langsame Komponente beisteuern - mit unterschiedlicher Beteiligung:
Links oben: Bei einem stark negativen Ruhepotential (hier ~-75
mV) beteiligen sich die NMDA-Rezeptoren kaum an der Entstehung des EPSP
(grün: Anteil AMPA, orange: Anteil NMDA, rot: Gesamtkurve des EPSP).
Rechts oben: Bei einem weniger stark negativen Ruhepotential (hier ~-40 mV) beteiligen sich zahlreiche NMDA-Rezeptoren am EPSP.
Links unten:
AMPA-Rezeptoren öffnen bei Anlagerung von Glutamat, unabhängig vom
Betrag des Ruhepotentials. Ist dieser hoch
(Registrierung links oben), bleiben die NMDA-Rezeptoren durch
Magnesiumionen blockiert.
Rechts unten:
Ist der Spannungsbetrag des Ruhepotentials gering (Registrierung rechts
oben), aktiviert Glutamat auch NMDA-Kanäle, die (außer Na+) auch Ca++ in die
Zelle lassen.
NMDA-Kanäle haben eine langsamere Kinetik als AMPA-Kanäle
AMPA-Glutamatrezeptoren

AMPA - Aminohydroxy-Methyl-Isoxazol-Propionsäure (Chemikalie)
Rezeptoren vom AMPA (α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid)-Typ (AMPAR) finden sich in den meisten exzitatorischen Synapsen des Gehirns, wo sie die Mehrzahl der L-Glutamat-Rezeptoren darstellen. AMPA-Rezeptoren steuern die rasche Komponente des Glutamateffekts (wenige Millisekunden) bei. Da sie sowohl für Na+ als auch K+ (annähernd gleich gut) durchgängig sind, liegt ihr Gleichgewichtspotential nahe beim Spannungs-Nullpunkt, d.h. es kommt vom Ruhepotential aus zu Depolarisation: Exzitatorische postsynaptische Potentiale (EPSPs).
Die Mehrzahl der AMPAR sind für Calciumionen undurchgängig (CI-AMPARs, calcium-impermeable AMPARs). Einige AMPAR (ihnen fehlt die GluA2-Untereinheit) sind hingegen - wenn geöffnet - gut für Ca++ durchgängig (CP-AMPARs, calcium-permeable AMPARs).
Kainat-Glutamatrezeptoren

Kainsäure (pflanzliches Analogon der Glutaminsäure)
Rezeptoren vom Kainat-Typ (Kainat - Salz der Kainsäure - zuerst in einer Meeresalge entdeckt, ein Strukturanalog der Glutaminsäure) beteiligen sich an glutamatbedingten EPSPs. Sie finden sich an
spezifischen Neuronentypen und auch an präsynaptischen glutamatergen
und GABAergen Axonterminals (wo sie die Transmitterfreisetzung
fördern).
Kainat-Rezeptoren sind (wie AMPA-Rezeptoren) durchgängig für Na+ und K+, sie wirken depolarisierend. Wegen ihrer Ähnlichkeit werden AMPA- und Kainat-Rezeptoren auch als Nicht-NMDA-Rezeptoren zusammengefasst.
NMDA-Glutamatrezeptoren

NMDA - N-Methyl-D-Aspartat (Aminosäurederivat)
Rezeptoren vom NMDA (N-methyl-D-aspartic acid)-Typ (NMDAR) sind für Kationen permeabel - nicht nur für Na+ und K+, sondern auch für Ca++ (das unterscheidet sie von CI-AMPARs), welches intrazelluläre Signalwege aktivieren kann. Der Einstrom von Calciumionen ist bedeutsam für synaptische Plastizität, Lernen und Gedächtnis.
Magnesiumionen blockieren bei höheren Beträgen des Membranpotentials den NMDA-Ionenkanal ("Magnesiumblock"), während Depolarisierung Mg++ aus dem Kanal in den Extrazellulärraum abdrängt und die Passage von Na+, K+ und Ca++ freigibt. Das heißt:
Extrazelluläre Magnesiumionen (auch andere zweiwertige Kationen)
blockieren den Ionenkanal, verlassen ihn aber bei Bindung von
Glutamat(agonisten).
NMDA-Kanäle haben eine langsamere Kinetik als AMPA-Kanäle (die Depolarisierung dauert bis zu etwa 100 ms), und ihr Öffnungsverhalten hängt nicht nur von der Bindung
des Agonisten ab, sondern auch von der Membranspannung; damit stellen sie eine Sonderform ionotroper Rezeptoren dar.
Ligandengesteuerte Coaktivierung von NMDA-Glutamatrezeptoren benötigt zusätzlich zu Glutamat / Aspartat auch Glycin oder Serin. Unterschiedliche Kombinationen von Isoformen solcher Bindungsstellen verleihen dem Rezeptor unterschiedliche Eigenschaften.
Der
Zustand der NMDA-Rezeptoren wird durch eine Vielzahl
psychoaktiver Drogen beeinflusst, Antagonisten können z.B. halluzinogen
wirken.
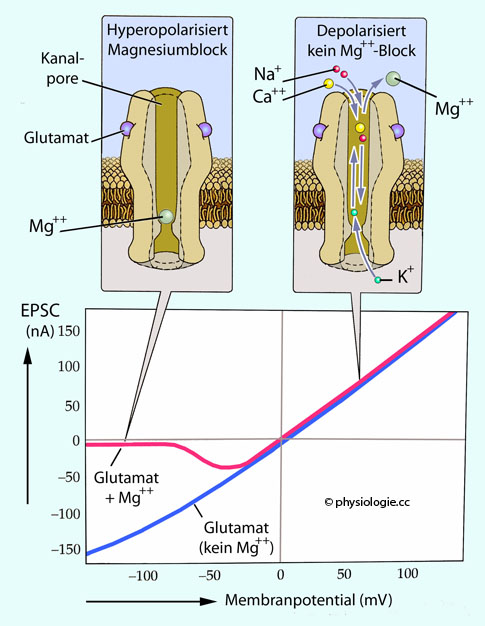
Abbildung: NMDA-Glutamatrezeptor
Nach
einer Vorlage in Augustine / Groh / Huettel / LaMantia / White (eds),
Neuroscience. Intl 7th ed. Oxford University Press 2024
Oben: Spannungsabhängige Blockade des Ionenkanals durch Mg++. Bei höherem Membranpotential / Hyperpolarisierung setzen sich Magnesiumionen in die Kanalpore und blockieren hier den Durchgang (links); Depolarisierung der Membran drängt Mg++ in den Extrazellulärraum ab und gibt die Pore für die Passage durch andere Kationen frei (rechts).
Unten: Exzitatorischer postsynaptischer Ionenstrom (EPSC, excitatoty postsynaptic current) abhängig vom postsynaptischen Membranpotential bei Mg++-blockierten (rote Kurve) und nicht blockierten glutamataktivierten NMDA-Ionenkanälen (blaue Kurve). Bei Membranpotentialwerten unter ~-25 mV macht sich der Magnesiumblock bemerkbar, der EPSC glutamataktivierter NMDA-Kanäle sistiert mit eingelagertem Mg++, ohne Mg++ nimmt der Kationeneinstrom stetig weiter zu
NMDA-Rezeptoren spielen eine wichtige Rolle für synaptische Plastizität und Lernfähigkeit. Ihre Steuerung über das Membranpotential ist eine Besonderheit, denn sie funktioniert nicht
(wie bei anderen spannungsgesteuerten Ionenkanälen) über eine
Konformationsänderung: Vielmehr werden bei einem Ruhepotential >70
mV Magnesiumionen durch die negative Ladung an der Membraninnenseite angezogen, lagern sich tief im Kanal an dessen Wand an und blockieren ihn. In diesem Zustand ist eine Anlagerung von
Glutamat nicht in der Lage, den Kanal zu öffnen.
Sinkt das
Ruhepotential unter etwa 60 mV (typischerweise über Aktivierung von AMPA- /
Kainat-Rezeptoren), erfolgt eine elektrostatische Abstoßung des Mg++; nun können Na+, K+ und Ca++
durch den Kanal diffundieren. Der Ionenkanal verhält sich gegenüber
diesen Ionen nichtselektiv, die Diffusion von Calciumionen ist aber
wegen des hohen Konzentrationsgefälles besonders intensiv. Intrazellulär steigt [Ca++] stark an, [Na+] und [K+] ändern sich hingegen kaum. Ca++
beeinflussr das Öffnungsverhalten verschiedener Ionenkanäle, aktiviert
zahlreiche Enzyme und die Transkription von Genen. Die Aktivierung intrazelluläree Signalwege führt u.a. zum Einbau zusätzlicher AMPA-Rezeptoren in die
postsynaptische Membran.
Der NMDA-Kanal ist also sowohl transmitter- als auch spannungsgesteuert. Die
Ionenströmung duch den NMDA-Rezeptorkanal ist am stärksten, wenn zwei
Faktoren zusammenkommen: Bindung des Transmitters (Glutamat) und
Depolarisierung der Membran. Insoferne kann der NMDA-Rezeptor als
"Koinzidenzdetektor" fungieren - wenn sowohl die präsynaptische als
auch die postsynaptische Zelle erregt ist.
Durch den relativ langsamen
Zeitverlauf des gesamten Vorgangs verlängern NMDA-Rezeptorkanäle weiters EPSPs oder IPSPs.
NMDA-Kanäle sind ionotrope Glutamatrezeptoren
|
Weiters werden als Cofaktoren Glycin oder Serin benötigt,
sowie die Anwesenheit von Zinkionen
(Abbildung oben). Serin ist eine Aminosäure, die von Nervenzellen als Neuromodulator verwendet wird:
Zwei der vier Untereinheiten des Glutamatrezeptors haben Bindungsstellen für Glutamat, zwei für Glycin und D-Serin
(Abbildung oben). Dabei existieren unterschiedliche Isoformen
der Glutamat- und der Serin-Bindungsstellen, die ontogenetisch
unterschiedlich kombiniert werden, sodass der Rezeptor abhängig von der
Entwicklungsphase und von der Lage im Gehirn entsprechend spezifische
funktionelle Eigenschaften aufweisen kann.
Zinkionen werden zusammen mit Glutamat in
präsynaptischen Vesikeln gespeichert und von dort freigesetzt; Serin
stammt aus Astrozyten, seine verstärkende Wirkung auf die
NMDA-Rezeptoren (über die Bindungsstellen für Glycin) könnte zur
Festigung von Engrammen beitragen.
Metabotrope Glutamatrezeptoren
Neben ionotropen gibt es auch G-Protein-gekoppelte metabotrope Glutamatrezeptoren (mGluR).
Diese produzieren schwächere postsynaptische Antworten als ionotrope, und
diese können nicht nur exzitatorisch, sondern auch (oft via Hemmung
postsynaptischer Calcium- und Natriumkanäle) inhibitorisch wirken - je
nach Umkehrpotential des jeweils regulierten Ions bzw. abhängig davon, ob sie Kanäle
öffnen oder schließen.

Abbildung: Aktivierung eines metabotropen Glutamatrezeptors (mGluR)
Nach einer Vorlage in Kandel / Koester / Mack / Siegelbaum (eds), Principles of Neural Sciences, 6th ed. 2021 (McGraw Hill)
mGluR sind heptahelikale metabotrope Rezeptoren (GPCR), die (als strukturelle Besonderheit) aus zwei identischen
Untereinheiten bestehen. Die Bindung von Glutamat führt zu einer Rotation
des membrangebundenen Rezeptoranteils und Aktivierung des G-Protein-Mechanismus,
der metabolische und
Ionenkanal-Aktivitäten beeinflusst (Effektor)
Metabotrope Glutamatrezeptoren werden aufgrund von
Unterschiedlichkeiten in Struktur, pharmakologischen Eigenschaften und
second-messenger-Signalwegen eingeteilt in
Gruppe I: Diese aktivieren die Kette
Phospholipase C → IP3 → Ca
++-Freisetzung, DAG → Proteinkinase C
Gruppe II und
III: Sie hemmen die Adenylylcyclase → cAMP sinkt.
Viele Transmitter
im Gehirn benutzen metabotrope Mechanismen, die auch durch
Medikamente beeinflussbar sind.
Glutamattransporter; dreiteilige Synapsen
Glutaminzyklus: In den Synapsenspalt freigesetztes Glutamat wird von Glutamattransportern
- diese finden sich an Nerven- und Gliazellen - aus dem
Extrazellulärraum entfernt, zu Glutamin verwandelt und von
der Nervenzelle wieder aufgenommen ("tripartite Synapsen", Abbildung).
Es gibt mehrere Glutamattransporter, die sich - z.B. durch Neuropharmaka - unterschiedlich beeinflussen lassen:
VGLUTs: vesicular glutamate transporters
EAATs: excitatory amino acid transporters
Glutamat-Cystein-Austauscher
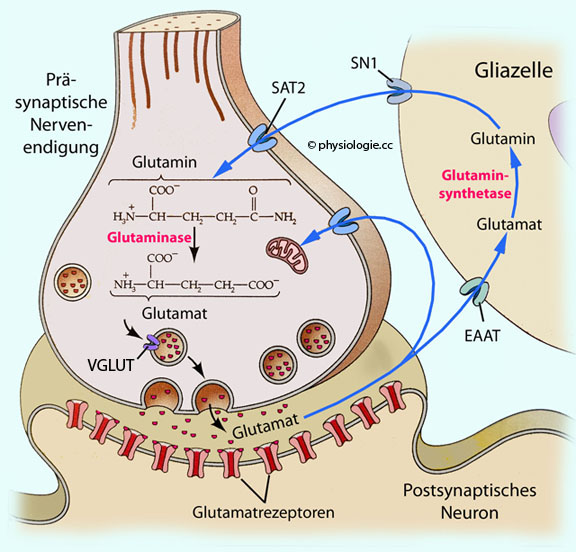
Abbildung: Dreiteilige (tripartite) Synapse und Glutamin-Glutamat-Zyklus
Nach
einer Vorlage in Augustine / Groh / Huettel / LaMantia / White (eds),
Neuroscience. Intl 7th ed. Oxford University Press 2024
Glutamat wird nach seiner Synthese im präsynaptischen Terminal in Vesikeln gespeichert (VGLUT = vesicular glutamate transporter). Nach seiner Freisetzung wird Glutamat von benachbartern Glia- und Nervenzellen via EAATs (excitatoty amino acid transporters) aufgenommen und aus dem synaptischen Spaltraum entfernt. Nach der Umwandlung zu Glutamin (Glutaminsynthetase) wird dieses über SN1-Transporter (SN steht für nukleophile Substitution) aus der Gliazelle und anschließend via System A-Transporter (SAT2) in die Endfortsätze präsynaptischer Neurone gebracht, wo es durch Glutaminase wieder zu Glutamat wird.
Gliazellen füllen fast den gesamten nicht-neuronalen Raum des Gehirns aus und unterstützen die Tätigkeit von Nervenzellen. Prä-
und postsynaptische Membranen können in unmittelbarer Nähe (Bruchteile eines µm) zu
Gliazellen positioniert sein. Astrozyten nehmen an sogenannten dreiteiligen Synapsen über Endfüßchen nicht nur Kaliumionen (die aktivierte Nervenzellen
vermehrt verlassen) aus dem Extrazellulärraum auf, sondern auch Transmitter (z.B. Glutamat), die Neuronen bei Erregung ebenfalls
freisetzen.
Astrozyten können auf diese Weise verschiedene Transmitter
zwischenspeichern und an Nervenzellen zurückgeben (Recycling): Außer Glutamat auch
Acetylcholin, GABA und zahlreiche weitere. Die Aufnahme geschieht meist
über metabotrope Rezeptoren, was oft auch das Membranpotential der
Gliazelle beeinflusst, zu vermehrtem Ca++-Einstrom und zum
Auftreten von "Calciumwellen" durch eine oder mehrere - miteinander
über gap junctions oder purinerge Signalübertragung
verknüpfte - Gliazelle(n) führen und unterschiedliche Reaktionen
nicht nur der Glia, sondern auch prä- und postsynaptischer Neurone
auslösen kann
Die Entfernung von Glutamat aus dem synaptischen Spaltraum reguliert den Erregungspegel des Nervengewebes.
Die Umwandlung
Glutamat → Glutamin erfolgt vor allem
in Gliazellen durch die Glutamat-Ammonium-Ligase (Glutaminsynthetase); die Umwandlung Glutamin → Glutamat hauptsächlich in Nervenzellen, durch Wirkung der
Glutaminase (dabei entsteht Ammoniak).
Glutamattransporter
nützen vor allem den Natriumgradienten (extrazellulär 140 mM,
intrazellulär 12 mM) zum Import von Glutamat (gegen dessen
Konzentrationsgefälle); so befördern sie 3 Na+-Ionen und ein Proton (H+) in die Zelle und ein K+-Ion
aus der Zelle. Ein Glutamatmolekül wird in diesem Fall also zusammen
mit insgesamt fünf anderen Ionen durch den Transporter bewegt (sodium and potassium coupled glutamate transporters).
Zerebrale Ischämie / Hypoxie
reduziert die Aktivität der Na/K-ATPasen, senkt den extrazellulären
Natrium- und den intrazellulären Kaliumspiegel. Damit nimmt auch die
Triebkraft für den Glutamat-Import ab, das von Nervenzellen
freigesetzte Glutamat reichert sich in der extrazellulären Flüssigkeit
an und wirkt exzitotoxisch.
Inhibitorische Neurotransmitter
GABA (γ-Aminobuttersäure)
und die Aminosäure Glycin
sind inhibitorische Neurotransmitter. Sie dienen sowohl direkter Hemmung
(durch Hyperpolarisierung) als auch zur Disinhibition, z.B. bei der
Bearbeitung von Sinnesreizen (Sensorik) oder im Bereich des Kleinhirns
und der Basalganglien (Motorik).
Unter Disinhibition versteht man die Abschwächung oder Unterdrückung einer hemmenden Aktivität, z.B. im Bereich der Basalganglien. Hier können dadurch "nachgeschaltete" anregende neuronale Muster freigegeben werden, z.B. von Bewegungsabfolgen im Thalamus.
Rezeptoren: Wie
Glutamatrezeptoren, sind auch GABA- und Glycinrezeptoren pentamer
aufgebaut. Ihre fünf Untereinheiten (bezeichnet als α, β, γ usw)
umgeben den zentralen Ionenkanal; jede davon hat eine große
extrazelluläre ligandenbindende Domäne, gefolgt von 4
membrandurchspannenden α-Helices (M1-M4).
Im Gegensatz zu den (sonst
ähnlich aufgebauten) nikotinischen Acetylcholinrezeptoren
sind ihre Ionenkanäle mit neutralen oder positiv geladenen basischen
Gruppen ausgekleidet, was ihre Spezifität für Anionen erklärt
(nikotinische Rezeptorenkanäle haben negativ geladene saure Gruppen).
GABA
 GABA wird durch Glutamat-Decarboxylase aus Glutamat gebildet und (wie
auch andere Transmitter) nach seiner Freisetzung teils abtransportiert
und abgebaut, teils wieder in die präsynaptische Zelle aufgenommen (reuptake) und wiederverwendet ("GABA-shunt", Abbildung).
GABA wird durch Glutamat-Decarboxylase aus Glutamat gebildet und (wie
auch andere Transmitter) nach seiner Freisetzung teils abtransportiert
und abgebaut, teils wieder in die präsynaptische Zelle aufgenommen (reuptake) und wiederverwendet ("GABA-shunt", Abbildung).
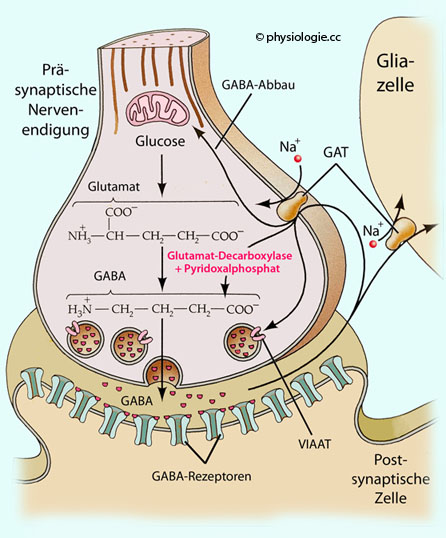
Abbildung: Bildung, Freisetzung und Wiederverwertung von GABA
Nach
einer Vorlage in Augustine / Groh / Huettel / LaMantia / White (eds),
Neuroscience. Intl 7th ed. Oxford University Press 2024
Wichtigster Ausgangsstoff für die GABA-Synthese ist Glucose (Glutamat aus dem Citratzyklus), weiters Pyruvat und Glutamin. Glutamat-Decarboxylase fördert die Umwandlung von Glutamat zu GABA (γ-Amino-Buttersäure), Pyridoxalphosphat (Vit. B6) ist dabei ein Cofaktor.
GABA wird nach seiner Synthese mittels VIAAT (vesicular inhibitory amino acid transporter)
- auch vesikulärer GABA-Transporter genannt - in präsynaptische Vesikel
verbracht, von wo es bei Erregung des GABAergen Neurons in den
synaptischen Spaltraum freigegeben wird.
Gliazellen
(Astrozyten) nehmen es hier via natriumabhängige Cotransporter - GAT,
GABA-Transporter (von denen es mehrere Arten gibt) - auf.
GAT wird auch von der präsynaptischenZelle exprimiert und sorgt für das
Recycling von GABA. Auch freigesetztes Glutamat wird in
Gliazellen aufgenommen, zu Glutamin verwandelt und von der Nervenzelle
wieder aufgenommen (Glutaminzyklus). Die Umwandlung Glutamat → Glutamin erfolgt durch die Glutaminsynthetase (hauptsächlich in der Gliazelle); die Umwandlung Glutamin → Glutamat durch die Glutaminase (hauptsächlich in der Nervenzelle).
Das meiste GABA wird schließlich zu Succinat abgebaut, das wiederum im Citratzyklus Verwendung findet
GABA ist der führende inhibitorische Transmitter
im Zentralnervensystem (Blockade der GABA-Synthese bewirkt
Krampfanfälle), vor allem an Interneuronen. Etwa 30% aller zerebralen
Synapsen arbeiten GABAerg; viele davon haben Peptid-Kotransmitter.
GABA
wirkt sowohl über ionotrope als auch über metabotrope Rezeptoren:
 Ionotrope Rezeptoren (GABAA) sind die im Körper am häufigsten vorkommenden GABA-Rezeptoren. Jeder
der pentameren Rezeptoren besteht aus Untereinheiten, deren Bauplan von
jeweils eigenen Genen codiert wird und aus deren Repertoire die
Rezeptoren unterschiedlich kombiniert sein können. Dieser "Bausteinset"
besteht aus
Ionotrope Rezeptoren (GABAA) sind die im Körper am häufigsten vorkommenden GABA-Rezeptoren. Jeder
der pentameren Rezeptoren besteht aus Untereinheiten, deren Bauplan von
jeweils eigenen Genen codiert wird und aus deren Repertoire die
Rezeptoren unterschiedlich kombiniert sein können. Dieser "Bausteinset"
besteht aus
Sechs Typen von α-Untereinheiten (GABRA1 bis GABRA6)
Drei Typen von β-Untereinheiten (GABRB1 bis GABRB3)
Drei Typen von γ-Untereinheiten (GABRG1 bis GABRG3)
Eine δ-Untereinheit (GABRD)
Eine ε-Untereinheit (GABRE)
Eine π-Untereinheit (GABRP)
Eine θ-Untereinheit (BABRQ)
Daraus ergibt sich eine enorme Zahl an
Kombinationsmöglichkeiten (Isoformen) mit spezifischen Eigenschaften. Beispielsweise
kann man alleine im Hippocampus etwa 20 verschiedene GABAerge Neuronen unterscheiden; die Kleinhirnrinde verfügt über verschiedene sehr spezielle GABAerge Zellen. Auch die Zahl der Substanzen, die pharmakologisch zur Beeinflussung von GABA-Rezeptoren verwendet werden, ist sehr groß.

Abbildung: GABA
A-Rezeptor (schematisch)
Nach einer Vorlage bei Jerrold S. Meyer / Linda F.
Quenzer, Psychopharmacology: Drugs, the Brain, and Behavior, 2nd Ed,
Sinauer Associates 2013
Der
Rezeptor verfügt über Bindungsstellen für den Transmitter
(Gamma-Aminobuttersäure) sowie Pharmaka wie Barbiturate, Benzodiazepine
oder neuroaktive Steroide (Geschlechtshormone)
Über GABAA-Rezeptoren
wirken Pharmaka, die sich nicht an die GABA-Bindungsstelle, sondern an
andere Rezeptorareale anlagern ( Abbildung)
und die Eigenschaften des Ionenkanals allosterisch verändern. Die Rezeptoren können ganz unterschiedliche
Empfindlichkeit für diese Modulatoren haben (die Untereinheiten des
Rezeptors können verschieden kombiniert sein). Benzodiazepine (z.B. Diazepam [Valium]) und Barbiturate (z.B. Phenobarbital) regen GABAA-Rezeptoren
an (Bildung von IPSPs), daher ihr
suppressiver Effekt auf das Gehirn. Diese Substanzen wirken
für sich alleine kaum, aber zusammen mit GABA auf den Rezeptor. Beispielsweise erhöhen
Benzodiazepine die Frequenz der Kanalöffnungen, Barbiturate und einige Steroide deren Dauer usw.
Eine Unterklasse GABA-gesteuerter Chloridkanäle, die ausschließlich aus ρ-Untereinheiten (3 Untertypen, ρ1 - ρ3) aufgebaut sind (die in GABAA-Rezeptoren nicht vorkommen), wurde früher als GABAC-Rezeptor bezeichnet und heißt jetzt GABAA-ρ-Rezeptor. Sie werden von Zellen der Netzhaut, in der Hypophyse, den colliculi superiores und im Rückenmark exprimiert und regieren intensiv auf GABA, nicht aber auf allosterische Modulatoren des Ionenkanals wie Barbiturate oder Benzodiazepine.
Postsynaptisch öffnen ionotrope GABA-Rezeptoren Chloridkanäle, rufen IPSPs hervor und wirken daher inhibitorisch, präsynaptisch wirken
sie als Autorezeptoren und hemmen die Transmitterfreisetzung.
GABAA-Rezeptoren öffnen Chloridkanäle und reduzieren die postsynaptische Erregbarkeit
|
Die Öffnung von Chloridkanälen bewirkt einen postsynaptischen Effekt, der vom aktuellen Membranpotential abhängt. Da das Gleichgewichtspotential für Chloridionen bei Nervenzellen bei -70 bis -80 mV liegt, hat die Öffnung von Cl--Kanälen
keinen Effekt, wenn das Membranpotential diesen Wert aufweist; ist es
niedriger, kommt es zu einer Erhöhung (hyperpolarisierend - IPSP), und
wenn es höher ist, zu einer Senkung des Membranpotentials
(depolarisierend - EPSP).
Der hemmende GABA-Effekt setzt demnach ein Membranpotential voraus, das
weniger als -70 bis -80 mV beträgt (Ruhepotential von Nervenzellen etwa
-65 mV). Hat das Membranpotential denselben Betrag wie das
Chlorid-Gleichgewichtspotential, verändert eine Öffnung von
Chloridkanälen das Membranpotential nicht, kann aber dennoch einen
inhibitorischen Effekt haben - in dem Sinne, dass sie den Beitrag am
Neuron wirkender depolarisierender Synapsen in Summe reduziert
(gewichtetes Mittel aller Synapseneinflüsse). Das Neuron wird weniger
depolarisiert, das Membranpotential stabilisiert.
Spontanaktive Zellen senken ihre Aktionspotentialfrequenz, wenn an ihnen Chloridkanäle geöffnet werden.
Es kann vorkommen, dass Neurone, die stark erregt sind, ihren intrazellulären Chloridspiegel verdoppeln. Das hebt das Cl--Gleichgewichtspotential an und kann zur Folge haben, dass die Öffnung von Chloridkanälen Cl- ausströmen lässt und die Zelle depolarisiert.
Metabotrope Rezeptoren (GABAB) finden
sich im Zentralnervensystem und im peripheren autonomen Nervensystem.
Sie funktionieren als Dimere (zwei septahelikale Untereinheiten B1 und
B2
sind über ihre intrazellulären Enden aneinander gekoppelt und müssen
kooperieren): GABA wirkt hier nur durch Bindung an B1, was eine
allosterische Veränderung an B2 mit Kopplung an G-Protein zur Folge hat.
GABAB-Rezeptoren
erhöhen über Gi die Offen-Wahrscheinlichkeit von K+-Kanälen (GIRKs) - das nähert das Membranpotential des Neurons an das Kalium-Gleichgewichtspotential an
senken die Offen-Wahrscheinlichkeit von Ca++-Kanälen via Reduktion der Aktivität der Adenylylcyclase.
Der Gesamteffekt ist eine Stabilisierung des Membranpotentials, Reduktion der Entladungsfrequenz und der Freisetzung des Neurotransmitters des betreffenden Neurons: GABAB-Rezeptoren haben einen inhibitorischen Effekt.
Auf GABAB-Rezeptoren wirkt eine Vielzahl von Agonisten (z.B. das muskelrelaxierende GABA-Analog Baclofen), Antagonisten und allosterisch wirksame Modulatoren.
Glycin
 Glycin
ist eine Aminosäure, die - vor allem in Rückenmark, Hirnstamm und in der Netzhaut - über Glycinrezeptoren wirkt, z.B. an motorischen Vorderhornzellen
(Renshaw-Selbsthemmung).
Glycin
ist eine Aminosäure, die - vor allem in Rückenmark, Hirnstamm und in der Netzhaut - über Glycinrezeptoren wirkt, z.B. an motorischen Vorderhornzellen
(Renshaw-Selbsthemmung).
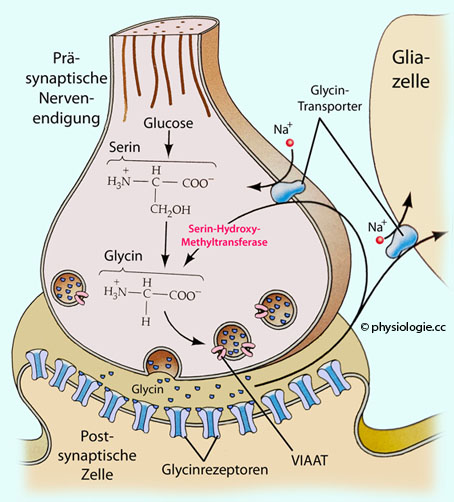
Abbildung: Synthese, Freisetzung und Recycling von Glycin
Nach
einer Vorlage in Augustine / Groh / Huettel / LaMantia / White (eds),
Neuroscience. Intl 7th ed. Oxford University Press 2024
Die neutrale Aminosäure Glycin kann auf mehreren Wegen synthetisiert werden; im Gehirn hauptsächlich aus der Aminosäure Serin. In
den synaptischen Spaltraum freigesetztes Glycin wird über spezifische
natriumabhängige Transporter (Cotransport mit Natrium) in die
präsynaptische Endigung retourniert, wo es - wie auch GABA - über VIAAT
(vesicular inhibitory amino acid transporter) in Vesikel verbracht wird.
Gliazellen exprimieren ebenfalls Glycintransporter und können Glycin aus dem Extrezellulärraum aufnehmen
Glycinerge
Synapsen sind im ZNS weniger stark verbreitet als GABA-Rezeptoren; im
Rückenmark machen sie etwa die Hälfte aller inhibitorischen Synapsen
aus.
Glycinrezeptoren sind aus vier α- (es gibt mehrere Typen von ihnen, die gemischt in die Rezeptoren eingebaut werden) und einer akzessorischen β-Untereinheit aufgebaut und wirken ausschließlich ionotrop. Glycin wirkt meist inhibitorisch, indem es (wie GABAA-Rezeptoren) Chloridkanäle öffnet ( Abbildung).
Bei Bindung von Glycin kommt es zu einer Konformationsänderung des
Rezeptors, die Ionenpore weitet sich, Chloridionen können in die Zelle
diffundieren und das postsynaptische Neuron wird hyperpolarisiert
(gehemmt).

Abbildung: Glycinrezeptor
Nach Beecham JE, Seneff S: The possible link between
autism and glyphosate acting as glycin mimetic - a revew of evidence
from gthe literature with analysis. J Mol Genet Med 2015; 9: 187
Das an inhibitorischen Synapsen essentielle Protein Gephyrin
vermittelt Clustering, Stabilisierung (für Interaktion mit dem
Zytoskelett) und Verfügbarkeit (Plastizität) von Glycin- und GABA-
Rezeptoren an der postsynaptischen Membran. Die Interaktion des Gephyrin mit anderen Proteinen ist komplex. An manchen GABAA-Rezeptoren ist Gephyrin nicht nachweisbar.
Eine Alpha-Einheit in dieser Darstellung vom Rezeptormolekül entfernt
Glycinrezeptoren öffnen Chloridkanäle und reduzieren die postsynaptische Erregbarkeit
|
Der Effekt ist abhängig vom Ausgangswert des Membranpotentials: das Gleichgewichtspotential der Chloridkanäle liegt bei ~-70 mV, d.h. ihre Öffnung führt bei Membranpotentialen unter diesem Wert zu Hyperpolarisation (IPSP's, inhibitorische postsynaptische Potentiale), bei Werten darüber hingegen zu Depolarisation.
Die Freisetzung von Glycin (wie auch von GABA) wird durch Tetanustoxin gehemmt, was die inhibitorische Glycinwirkung an betreffenden Synapsen konterkariert und Muskelkrämpfe auslöst.
Glycinrezeptoren können durch das pflanzliche Alkaloid Strychnin blockiert werden, was ebenfalls Muskelkontraktionen enthemmt (Strychnin gilt als Krampfgift).
Glycin bindet als Co-Agonist des Glutamats auch an eine spezielle Sequenz des NMDA-Rezeptors und verstärkt so die exzitatorische Neurotransmission.
Chloridabhängigkeit der Synapsenwirkung:
Der hyperpolarisierende (inhibitorische) Effekt der GABA- und
Glycin-Rezeptoren hängt vor allem von der Öffnung von Chloridkanälen
ab. Die intrazelluläre Chloridkonzentration ist allerdings ziemlich
variabel; steigt sie in der Nervenzelle an, kann sich der Chloridstrom
durch die Cl--Kanäle umkehren (Ausstrom statt Einstrom), und
die Aktivierung des Chloridkanals depolarisiert die Zelle statt sie zu
hyperpolarisieren. Deshalb ist die Aktivität von K+/Cl--Kotransportern (KCC) wichtig: Sie nutzen den Kaliumgradienten für die Entfernung von Chloridionen aus den Neuriten.
Die Öffnung von Ionenkanälen (wie GABA- oder Glycin-betriebenen Chloridkanälen) senkt auch den Membranwiderstand und damit die Längskonstante der Membran. Das verringert die Reichweite der elektrotonischen Übertragung örtlicher Depolarisationen (EPSPs)
und damit deren Effektivität für die Bildung von Aktionspotentialen.
Mit anderen Worten, offenstehende Ionenkanäle verringern die
Erregbarkeit der betreffenden Membranstelle; die Öffnung von Glycin-
oder GABA-Kanälen wirkt auch auf diese Weise erregungsdämpfend (shunting inhibition).
 Weitere inhibitorische Neurotransmitter sind ß-Alanin und Taurin.
Weitere inhibitorische Neurotransmitter sind ß-Alanin und Taurin.
Über Rezeptoren für
Serotonin, Noradrenalin, Dopamin und
Acetylcholin im Gehirn s.
dort
Präsynaptische Bahnung
(presynaptic facilitation) steigert die Menge an freigesetztem Transmitter; so nimmt der postsynaptische Effekt bei wiederholter Reizung (z.B. bei
einer Frequenz von 20/s) zu (Anwachsen des postsynaptischen
Potentials). Dem Effekt liegt eine Steigerung des Ca++-Einstroms in die präsynaptischen Endigungen - durch spannungs- oder ligandengesteuerte Calciumkanäle oder aus dem endoplasmatischen Retikulum - zu Grunde. Erhöhtes [Ca++] vermehrt die Freisetzung von Transmitter aus Vesikeln. Hört die Reizung auf, wird Ca++ innerhalb von Sekunden wieder aus dem Zytosol entfernt (mittels Ca++-ATPase in das Retikulum, mittels Na+/Ca++-Austauscher in den Extrazellulärraum).
Im Gegensatz dazu können postsynaptische Potenzierungen über längere Zeit anhalten (vgl. dort).
Colokalisation und Cotransmission
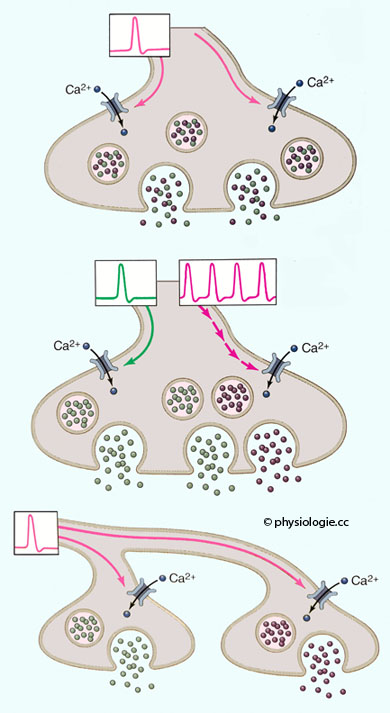
Abbildung: Gleichzeitige Freisetzung mehrerer Transmitterstoffe
Nach einer Vorlage bei Guyton and Hall, Textbook of Medical Physiology, 15th ed. Elsevier 2026
Oben:
Gemeinsame Freisetzung (co-release). Die verschiedenen Transmitterstoffe können in ein und denselben
Vesikeln gespeichert vorliegen und werden dann automatisch gemeinsam
freigesetzt.
Mitte: Cotransmission und unterschiedliche Calciumsensitivität. Die verschiedenen Transmitterstoffe können im selben Neuron in verschiedenen Vesikelpopulationen
vorliegen. Ihre Freisetzung kann dann spezifisch erfolgen, und zwar
über unterschiedliches Ansprechverhalten gegenüber der intrazellulären
Konzentration von Calciumionen, die bei Erregung der Zelle mit
unterschiedlichen Aktionspotentialfrequenzen auftreten kann.
Unten:
Cotransmission bei räumlicher Trennung. Unterschieldiche Axonterminale (Endknöpfchen) können unterschiedliche
Transmitter speichern, die Regulierung der entsprechenden
Transmitterfreigabe erfolgt auf lokaler Ebene
Kleinmolekulare
(rasch wirksame) Transmitter (wie Glutamat, Serotonin etc) und (langsam
wirkende) Neuropeptide finden sich häufig in Neuronen gleichzeitig.
Ihre präsynaptische Endigungen (Axonterminals) enthalten häufig zwei oder mehrere Transmitterarten (Colokalisation), die bei Erregung des Neuriten gleichzeitig freigesetzt werden (Cotransmission, Abbildung).
Als Cotransmission bezeichnet
man die gemeinsame Freisetzung verschiedener Neurotransmitter von ein
und demselben axonalen Nerventerminal. Beispiele sind
 Glutamat mit Dopamin
Glutamat mit Dopamin
Glutamat mit Acetylcholin
Glutamat und Dynorphin (Hippocampus)
GABA mit Glycin
Acetylcholin mit Calcitonin
Acetylcholin mit Vasopressin
Wie eine "Senderzelle" die Freisetzung verschiedener Transmitterstoffe in differenzierter Weise (selektiv) vornehmen kann, zeigt nochmals die folgende Abbildung:
Abbildung: Selektive Freisetzung colokalisierter Transmitter
Nach einer Vorlage in Boron / Boulpaep: Concise Medical Physiology, Elsevier 2021
Mitte:
Ein präsynaptisches Terminal, das in kleinen Vesikeln einen
kleinmolekularen Transmitter und in großen Vesikeln ein Neuropeptid
speichert. Große Vesikel sind weiter vom synaptischen Spalt (und den postsynaptischen aktiven Zonen) entfernt als kleine.
Oben: Niedrigfrequente Reizung
(Aktionspotentiale in größerem zeitlichem Abstand, s. Inset) führt zu
geringgradigem Calciumeinstrom, der nur auf kleine (synapsenspaltnahe)
Vesikel wirkt. Der kleinmolekulare Transitter wird in den synaptischen
Spalt abgegeben.
Unten: Hochfrequente Reizung (Aktionspotentiale
in geringem zeitlichem Abstand, s. Inset) führt zu ausgedehntem Anstieg
der
Calciumkonzentration, der sich nun auch auf große Vesikel auswirkt, aus
denen das Neuropeptid in den Extrazellulärraum freigesetzt wird
Reizstärke und Exozytose: Die Erregung eines Axons führt zur Öffnung von Calciumkanälen in seiner präsynaptischen Membran und damit zu Ca++-Einstrom
(Abbildung). Abhängig von der Intensität des Calciumanstiegs im
Axonterminal können ein oder mehrere Vesikel ihren Inhalt (Transmitter)
in den Extrazellulärraum entleeren. Aber nicht jedes Aktionspotential
steigert den intrazellulären Calciumspiegel ausreichend stark, um den
Exozytosemechanismus zu triggern. Einzelne präsynaptische Entladungen
können also auch wirkungslos auf das Membranopotential des
postsynaptischen Neurons bleiben.
Nimmt die Zahl der präsynaptischen Erregungen zu, steigt auch die
Wahrscheinlichkeit der Transmitterfreisetzung. Kleine Vesikel (etwa 40
nm Durchmesser, nahe am synaptischen Spalt gelegen) exozytieren ihren
kleinmolekularen Transmitter (z.B. Glutamat) schon bei niedrigfrequenter Reizung. Große (100-200 nm), weiter vom Synapsenspalt entfernte, mit Neuropeptiden beladene erst bei hoher Aktionspotentialfrequenz (Abbildung).
So hat die Nervenzelle eine gewisse Kontrolle über das Muster der
freigesetzten Transmitter - über die Ausdehnung der durch die
Erregungspulse im Terminal freigesetzten Calciumwolken: Je höher die
Aktionspotentialdichte, desto stärker in das Terminal hinein dringen
freie Calciumionen, und desto eher triggern diese die Aktivierung
großer Vesikel.
Viele Cotransmitter
wirken als Neuromodulatoren und sind meist Neuropeptide (Somatostatin,
Substanz P, Angiotensin II, Enkephaline..), aber auch ATP, NO u.a. - s.
oben. Ihre Aufgabe ist die Veränderung der
Intensität und Dauer des postsynaptischen Effekts des "klassischen"
Transmitterstoffs. Für ihre (Ca++-getriggerte)
Freisetzung aus Speichervesikeln braucht es eine intensivere
Depolarisierung der (präsynaptischen) Nervenzelle als für die
Freisetzung des "primären" (niedermolekularen) Transmitters alleine.
Konvergenz und Divergenz
Auf eine
Nervenzelle wirken zahlreiche andere synaptisch ein (Konvergenz), andererseits schaltet jedes Neuron mit den Verzweigungen seines Axons auf zahlreiche andere (Divergenz) - Größenordnung jeweils bis etwa 5.104.
Abbildung: Nervenzelle, Kon- und Divergenz
Konvergenz: Zu einer Zelle läuft Information von mehreren anderen Zellen zusammen
Divergenz: Eine Zelle
sendet Information an mehrere andere Zellen
Die logischen Verschaltungen zwischen den Zellen erfolgen über
Synapsen. Nervenzellen funktionieren wie
Rechenmaschinen, die nach der Logik der Summation (räumlich und zeitlich) synaptischer Einflüsse
funktionieren.
Konvergenz und Divergenz
sind Verschaltungsprinzipien, welche Grundlage komplexer
Funktionsmuster sind. Man findet sie auf unterschiedlichen Ebenen (Abbildungen). So
können Neurotransmitter einerseits auf verschiedene Rezeptoren (und deren Folgemechanismen) zugreifen (Divergenz),
andererseits können unterschiedliche Transmitter über ihre jeweiligen
Rezeptoren denselben Mechanismus (G-Protein, second messender,
zellulären Effekt) anregen (Konvergenz).

Abbildung: Konvergenz- und Divergenzprinzip neuronaler Verschaltung
Nach einer Vorlage bei Silverthorn, Human Physiology, an integrated approach, 4th Int'l ed. 2007, Pearson / Benjamin Cummings
Konvergenz: Nervenzellen
empfangen Impulse von mehreren anderen Neuronen. Dadurch können z.B.
schwache Impulse summiert und überschwellig, oder Assoziationen
verfestigt werden (Koinzidenz).
Divergenz:
Nervenzellen
senden über ihre Kollateralen Impulse an mehrere andere. Dies
ermöglicht breite Streuung neuronaler Information und z.B.
Bahnungseffekte in Nachbarneuronen
Beispiele:
Divergenz: Noradrenalin wirkt auf α
1-Rezeptoren (über
G-Protein, PLC, PKC), α
2-Rezeptoren
(über G-Proteine und z.T. PLC, PKC), und ß-Rezeptoren (über G-Protein,
Adenylylcyclase, cAMP) ganz unterschiedlich auf diverse Ionenkanäle und
damit auf das Membranpotential
Konvergenz: Kaliumkanäle werden über Gα-Protein
von Rezeptoren für Adenosin, Acetylcholin, Dopamin, Enkephalin, GABA,
Noradrenalin, Serotonin und Somatostatin beeinflusst
Konvergenz und Divergenz spielen weiters für die sinnesphysiologische Analyse
von Umweltreizen eine wichtige Rolle. So werden über afferente Bahnen
heranströmende Sinnesmeldungen im ZNS abstrahiert, Kontraste
verstärkt, Muster erkannt und Merkmale zugeordnet.
Aktionspotentiale von mehreren Rezeptorzellen in einem Sinnesorgan
konvergieren auf einzelne nachgeschaltete Nervenzellen, gleichzeitig
divergiert Information von einer Rezeptorzelle auf mehrere Neuronen.
Diese Verschaltungen unterliegen modifizierenden Einflüssen durch
Interneurone und deszendierende Impulse. Auf diese Weise können aus der
anflutenden sensorischen Information Gestalten herausgearbeitet werden,
die in der jeweiligen Situation besonders bedeutsam sind.
Das Gebiet, das jeweils zu
einer zentralen Nervenzelle konvergiert, heißt rezeptives Feld.
Rezeptive Felder überschneiden sich, weil Rezeptorzellen mehreren
rezeptiven Feldern zugeordnet sind (Divergenz).
Chemische Synapsen haben “Einbahnwirkung” (Ausnahme gap junctions), sofern sie
nur Einfluss in eine Richtung - prä- auf subsynaptisch - zulassen. Sie
haben Bahnungs- oder Hemmfunktion durch Herabsetzung oder Steigerung
des subsynaptischen Membranpotentials.
Gedächtnisfunktion entsteht, wenn wiederholte Aktivierung von Synapsen ihre Wirkung abschwächt
(Gewöhnung, Habituation) oder verstärkt (synaptische Potenzierung). Dies ist bei GABAergen Synapsen der Fall.
Nervenzellen
wirken zusammen (Kooperation, Assoziation; Langzeitpotenzierung über viele Stunden).
Die Tatsache, dass sich die Stärke von Synapsenwirkungen
funktionsabhängig ändert, nennt man synaptische Plastizität; sie ist
eine Grundlage für Lernprozesse.
Zur Physiologie der Glia s. dort
SNARE-Proteine
(s. oben)
können durch Proteasen beschädigt werden, die von pathogenen
Bakterien (Clostridium tetani → Tetanospasmin, Wundstarrkrampf; Clostridium botulinum →
Botulinumtoxin, Lebensmittelvergiftung - lat. botulus = Wurst) erzeugt werden. Dadurch wird der Mechanismus der
Transmitterfreisetzung gestört und es treten
Wundstarrkrampf (Tetanus
im pathologischen Sinn: Hemmung inhibierender Synapsen - GABA, Glycin - an Renshaw-Zellen) oder
Lähmungen (Acetylcholin an exzitatorischen Synapsen bzw. der motorischen Endplatte
wird nicht freigesetzt) auf (Botulismus).
Schutz bietet eine Impfung gegen
Wundstarrkrampf (evt. simultan: Aktiv und passiv gleichzeitig).
Personen, welche die Kontrolle über ihre Atemmuskeln verlieren, müssen künstlich beatmet werden.

 Neuronen sind über Synapsen miteinander
verknüpft. Der präsynaptische Teil setzt bei Erregung Transmitter
frei, der postsynaptische trägt Rezeptormoleküle. Der
Transmitter wird nach seiner Freisetzung wiederaufgenommen und/oder
abgebaut, er kann auch präsynaptisch negativ rückkoppelnd oder
über den Kreislauf neuroendokrin wirken (längere Halbwertszeit). Neurotransmitter sind z.B. Glutamat / Aspartat,
GABA, Glycin, Acetylcholin, Katecholamine, Serotonin; Kotransmitter Peptide, Purine, NO,
Eikosanoide Neuronen sind über Synapsen miteinander
verknüpft. Der präsynaptische Teil setzt bei Erregung Transmitter
frei, der postsynaptische trägt Rezeptormoleküle. Der
Transmitter wird nach seiner Freisetzung wiederaufgenommen und/oder
abgebaut, er kann auch präsynaptisch negativ rückkoppelnd oder
über den Kreislauf neuroendokrin wirken (längere Halbwertszeit). Neurotransmitter sind z.B. Glutamat / Aspartat,
GABA, Glycin, Acetylcholin, Katecholamine, Serotonin; Kotransmitter Peptide, Purine, NO,
Eikosanoide
Symmetrische
Synapsen haben gleich große prä- und postsynaptische Zonen (meist
inhibitorisch), bei asymmetrischen ist der postsynaptische Apparat
ausgeprägter (meist exzitatorisch). Man unterscheidet axodendritische,
axosomale, axoaxonale, auch dendrodendritische Synapsen. Die
Signalübertragung kann Millisekunden (Glutamat, GABA, Glycin, Acetylcholin nikotinerg), Sekunden (Katecholamine, Acetylcholin
muskarinerg), oder bis Tage wirken (Neuromodulation).
Fazilitation bedeutet Erhöhung, Depression Erniedrigung der
synaptischen Wirkung nach hochfrequenter Reizung des betreffenden
Synapsensystems (synaptische Plastizität),
erklärbar durch Verstärkung oder Abschwächung der
Transmitterfreisetzung und der Rezeptoransprechbarkeit
Einzelne synaptische Depolarisationen (EPSPs) depolarisieren um 0,01-1 mV, sie bleiben alleine unterschwellig. Mehrere EPSPs können das Membranpotential über das Schwellenpotential hinaus reduzieren (Summation) und so ein Aktionspotential auslösen. Die
Summation kann zeitlich (d.h. nacheinander) oder räumlich (d.h.
gleichzeitig durch mehrere Synapsen) erfolgen. Bei zeitlicher Summation
ist die [Ca++] im präsynaptischen Zytoplasma durch initiale
Erregung erhöht, das verstärkt die Transmitterfreisetzung sowie knapp
nachfolgende EPSPs. Inhibierende Synapsen
hyperpolarisieren meist durch erhöhte Durchlässigkeit für Chloridionen
oder Anstieg der Kaliumdurchlässigkeit. IPSPs stabilisieren das
Membranpotential
Neurotransmitter
und Neuropeptide werden vesikulär gespeichert: Protonenpumpen stellen einen
pH-Wert von ~5,4 ein, Transporter konzentrieren den Transmitter im
Vesikel. Die Transmitterfreisetzung erfolgt nach folgender Sequenz: Depolarisierung öffnet spannungsgesteuerte Ca++-Kanäle → Exozytose (elektro-sekretorische Kopplung), über Calmodulin und Proteinkinasen Aktivierung weiterer Vesikel → Transmitterfreisetzung.
Zahlreiche spezielle Proteine sind involviert (Synaptotagmin,
Synaptobrevin, Syntaxin, SNARE-Proteine). Jedes
Aktionspotential führt zur Entleerung hunderter Vesikel (Freisetzung von einigen 104 Transmittermolekülen)
Transmitter binden an unterschiedliche Rezeptor-Subtypen (verschiedene Effekte möglich). Rezeptoren
verändern ihre Ansprechbarkeit gegenüber dem Neurotransmitter: Phosphorylierung / Dephosphorylierung, Wechsel
rezeptorbeladener Membranabschnitte zwischen Zellmembran und
Zellinnerem. Drei
Mechanismen beenden die Transmitterwirkung: Sinkende Konzentration
durch Diffusion in das umliegende Interstitium, enzymatischer Abbau im
synaptischen Spalt, Aufnahme in präsynaptische Neurone und Gliazellen. Praktisch alle
Stufen des synaptischen Wirkmechanismus können pharmakologisch /
toxikologisch beeinflusst werden
Glutamat ist
der wichtigste exzitatorische Transmitter (~50% aller zerebralen Synapsen), es ist mit dem Zitratzyklus verknüpft und kann zu Glutamin
amidiert werden. Glutamat wirkt
über vier Rezeptorklassen - eine metabotrope: mGluR, drei ionotrope: AMPA-, NMDA-,
Kainat-R. mGluR
werden eingeteilt in Gruppe I: Aktivierung Phospholipase C → IP3 →
Ca++-Freisetzung, DAG → Proteinkinase C; und Gruppe II und III: Hemmung
der Adenylylcyclase → cAMP sinkt. AMPA sind durchgängig für Na+ und K+, sie finden sich an den meisten exzitatorischen Synapsen und bewirken rasche EPSPs. Der NMDA-Glutamatrezeptor ist ein Na+-, K+- und Ca++-Kanal, für seine Öffnung reicht die Bindung von Glutamat nicht aus: Mg++
blockiert den Ionenkanal, Depolarisierung entfernt es, z.B. durch
Aktivierung von AMPA- oder Kainat-Rezeptoren; als Cofaktoren wirken
Glycin oder Serin sowie Zinkionen, die zusammen mit Glutamat aus
präsynaptischen Vesikeln freigesetzt werden. Kainat-Rezeptoren lassen Na+ und K+ passieren
Nerven- und Gliazellen nehmen Glutamat über Transporter auf - dabei gelangen 3 Na+ und ein H+ in die, ein K+ aus der Zelle. So wird Glutamat aus dem synaptischen Spaltraum entfernt und der Erregungspegel des Nervengewebes reguliert (Schutz vor Exzitotoxizität: diese kann bei Ischämie durch verringerte Glutamatclearance auftreten). Gliazellen wandeln freigesetztes Glutamat zu Glutamin um (Glutamat-Ammonium-Ligase), Nervenzellen nehmen dieses auf und wandeln es in Glutamat um (Glutaminase), dabei entsteht Ammoniak (das aus dem Gehirn
entfernt wird)
GABA und Glycin sind inhibitorische Neurotransmitter; beide werden präsynaptisch vesikulär gespeichert. Glutamatdecarboxylase bildet GABA (γ-Aminobuttersäure) aus Glutamat; GABA ist der führende inhibitorische Transmitter im Zentralnervensystem (~30% aller zerebralen Synapsen). Astrozyten
nehmen GABA und Glutamat auf und stellen dem präsynaptischen Neuron
Glutamin für die Transmittersynthese zur Verfügung (Glutaminzyklus). GABA wirkt über drei Rezeptor-Haupttypen: Ionotrope sind die häufigsten (GABAA), sie öffnen Chloridkanäle und reduzieren die postsynaptische Erregbarkeit (IPSP); metabotrope (GABAB) hemmen die Adenylylcyclase, öffnen K+- und schließen Ca++-Kanäle, beides stabilisiert das Membranpotential; und Chloridkanäle (GABAC: Retina, Rückenmark, colliculi superiores, Hypophyse). GABA wird nach seiner Freisetzung teils abgebaut, teils in präsynaptische Neuronen aufgenommen. - Glyzin öffnet an seinen Rezeptoren Chloridkanäle; der Effekt ist abhängig vom Ausgangswert des Membranpotentials (Cl-- Gleichgewichtspotential ~-70 mV, bei geringerem Wert des Membranpotentials → IPSP, bei höherem → EPSP) und der Öffnung der Chloridkanäle
Wird das
Membranpotential eines Axons durch aufgeschaltete Neurite verändert,
beeinflusst das die Menge des von ihm (aktionspotentialbedingt)
freigesetzten Transmitters. Dieses Prinzip der präsynaptischen Hemmung ist insbesondere im Rückenmark wirksam.
Neuromodulation ist die prä- oder postsynaptische, indirekte
Beeinflussung der Freisetzung oder Wirkung von Transmittern - meist
über veränderte Permeabilität von K+- und Ca++-Kanälen. Neuromodulation erfolgt über Kotransmitter, wirkt längerfristig und ist an Gedächtnisprozessen beteiligt
Mehrere tausend synaptische Endigungen von verschiedenen anderen Neuronen können auf
eine einzelne Nervenzelle einwirken (Konvergenz), andererseits wirkt
ein Neuron auf mehrere andere ein (Divergenz). Konvergenz und Divergenz ermöglichen die Analyse und Beeinflussung von Erregungsmustern: Sinnesmeldungen werden abstrahiert,
Kontraste verstärkt, Muster erkannt, Merkmale zugeordnet. Schwache
Impulse können summiert und überschwellig, Assoziationen
verfestigt werden (Koinzidenzdetektion). Das Gebiet in einem Sinnesorgan, das zu
einer zentralen Nervenzelle konvergiert, nennt man dessen rezeptives
Feld. Rezeptive Felder überschneiden sich (Divergenz)
|
 Die Informationen in dieser Website basieren auf verschiedenen Quellen:
Lehrbüchern, Reviews, Originalarbeiten u.a. Sie
sollen zur Auseinandersetzung mit physiologischen Fragen, Problemen und
Erkenntnissen anregen. Soferne Referenzbereiche angegeben sind, dienen diese zur Orientierung; die Grenzen sind aus biologischen, messmethodischen und statistischen Gründen nicht absolut. Wissenschaft fragt, vermutet und interpretiert; sie ist offen, dynamisch und evolutiv. Sie strebt nach Erkenntnis, erhebt aber nicht den Anspruch, im Besitz der "Wahrheit" zu sein.
Die Informationen in dieser Website basieren auf verschiedenen Quellen:
Lehrbüchern, Reviews, Originalarbeiten u.a. Sie
sollen zur Auseinandersetzung mit physiologischen Fragen, Problemen und
Erkenntnissen anregen. Soferne Referenzbereiche angegeben sind, dienen diese zur Orientierung; die Grenzen sind aus biologischen, messmethodischen und statistischen Gründen nicht absolut. Wissenschaft fragt, vermutet und interpretiert; sie ist offen, dynamisch und evolutiv. Sie strebt nach Erkenntnis, erhebt aber nicht den Anspruch, im Besitz der "Wahrheit" zu sein.





 Abbildung: Präsynaptische Hemmung (presynaptic inhibition)
Abbildung: Präsynaptische Hemmung (presynaptic inhibition)

