Diabetes mellitus: διαβαίνειν = hindurchfließen, mel = Honig (mellitus = honigsüß)
Diabetes mellitus: διαβαίνειν = hindurchfließen, mel = Honig (mellitus = honigsüß)| Insulin
wirkt über Tyrosinkinase-Rezeptoren am Zielgewebe (vor allem
Fett-, Muskel- und Leberzellen) je nach Enzymausstattung
unterschiedlich: In der Leber fördert es die Synthese von Glykogen und
Fett; im Muskel die Protein-, im Fettgewebe die Lipogenese, in beiden
die Aufnahme von Glucose über den Einbau entsprechender Transporter
(GLUT-4) in die Zellmembran. Zahlreiche Signale regen die Insulinsekretion in den B-Zellen des Pankreas an: Erhöhung des Glucose-, Amino- und Fettsäurespiegels im Blut; Aktivität autonomer Nerven (sympathisch, parasympathisch); vermehrte Hormonkonzentrationen (Inkretin-Effekt durch Gastrin, Sekretin u.a.), einschließlich des Insulins selbst (autokrines Feedback). Die Anregung der B-Zelle durch Glucose erfolgt so: GLUT-2-Transporter lassen Glucose in die Zelle, ATP wird vermehrt gebildet und sein Spiegel steigt an. Das blockiert ATP-sensitive Kaliumkanäle und reduziert den K+-Ausstrom - die resultierende Depolarisierung führt zu Einstrom von Ca++-Ionen und Freisetzung des in Vesikeln gespeicherten Hormons. Dieser Vorgang erfolgt nicht kontinuierlich, sondern pulsatil (alle 3-6 Minuten) - so lange dauert auch die biologische Halbwertszeit des Insulins, das auf diese Weise wirksam bleibt (kontinuierliche Anwesenheit des Hormons führte zu receptor downregulation, die Zelle wäre refraktär, der Signalweg blockiert). Ein herausragender Insulineffekt ist die Senkung des Blutzuckerspiegels - Glucose wandert aus dem extrazellulären Raum in die Zellen. Die Glucosekonzentration im Blutserum sollte nüchtern (postabsorptiv) 3,3-6,0 mM (60-110 mg/dl) betragen. Erniedrigte Glucosewerte (Hypoglykämie) gefährden die Funktion primär glucoseabhängiger Gewebe, insbesondere des Gehirns. Erhöhung des Blutzuckerspiegels (Hyperglykämie) ist nach Mahlzeiten physiologisch, weil resorbierter Zucker zunächst ins Blut gelangt. Der darauf erfolgende Insulinanstieg senkt den Glucosespiegel rasch wieder in den Referenzbereich. Im Gehirn hat Insulin Signalwirkung im Sinne eines Sättigungssignals (es wird bei Zuckerzufuhr ausgeschüttet) und beeinflusst ausser Essverhalten, Blutzuckerregulation, Energiehaushalt und Körpergewicht auch Bewusstsein und Gedächtnisbildung. Glucose kann sich mit anderen Biomolekülen verbinden, Komplexe bilden und - bei chronisch erhöhten Werten - längerfristig degenerativ wirken (Durchblutungsstörungen, Nervenschäden bei chronischem unbehandeltem Diabetes). |
 Entdeckung Zentralnervöse Wirkungen Bildung und Abbau Inkretinwirkung Normalwerte Steuerung, (pulsatile) Freisetzung Insulinrezeptor Metabolische Wirkungen
Entdeckung Zentralnervöse Wirkungen Bildung und Abbau Inkretinwirkung Normalwerte Steuerung, (pulsatile) Freisetzung Insulinrezeptor Metabolische Wirkungen

 Inkretine Insulinempfindlichkeit
Inkretine Insulinempfindlichkeit
 Core messages
Core messages
 Abbildung: 24-Stunden-Profil des Insulinspiegels einer gesunden Person
Abbildung: 24-Stunden-Profil des Insulinspiegels einer gesunden Person Zellen nehmen Glucose über erleichterten Transport via Glucosetransporter auf, die sich in ihrer Zellmembran befinden (sie müssen diese Transporter exprimieren).
Treibende Kraft des transmembranalen Glucosestroms ist das
Konzentrationsgefälle - in der extrazellulären Flüssigkeit 4-6 mM
(postprandial höher), im Zytosol (technisch schwierig bestimmbar) 0,4-6
mM. Üblicherseise ist der Konzentrationsgradient in die Zelle hinein
gerichtet. Nehmen Zellen Glucose auf, trägt das zu einer Senkung des
Blutzuckerspiegels bei, weil sie auf diese Weise Glucose aus dem
Extrazellulärraum entfernen.
Zellen nehmen Glucose über erleichterten Transport via Glucosetransporter auf, die sich in ihrer Zellmembran befinden (sie müssen diese Transporter exprimieren).
Treibende Kraft des transmembranalen Glucosestroms ist das
Konzentrationsgefälle - in der extrazellulären Flüssigkeit 4-6 mM
(postprandial höher), im Zytosol (technisch schwierig bestimmbar) 0,4-6
mM. Üblicherseise ist der Konzentrationsgradient in die Zelle hinein
gerichtet. Nehmen Zellen Glucose auf, trägt das zu einer Senkung des
Blutzuckerspiegels bei, weil sie auf diese Weise Glucose aus dem
Extrazellulärraum entfernen. Insulin
regt Zellen, die über Insulinrezeptoren verfügen (primäre Ziele: Adipozyten,
Muskelzellen, Leberzellen) zur Glucoseaufnahme an und kann so den
Blutzuckerspiegel senken. GLUT4-Permeasen sind insulinabhängig (zu Glucosepermeasen s. dort). Epithelzellen in Nieren (Tubuli) und Darm (Mucosa) sowie Nervenzellen exprimieren keine Insulinrezeptoren und sind nicht
insulinabhängig. Andernfalls könnten sie im Hungerzustand keine Glucose
aufnehmen; ihre Glucoseaufnahme ist auch im postresorptiven Zustand gewährleistet (sie verfügen über nicht-insulinabhängige Permeasen für Glucose wie GLUT2, GLUT3 u.a. In Gefahren- bzw. Belastungssituationen wird die Insulinausschüttung gehemmt (Näheres s. dort). Bei solchen fight-or-flight-Situationen
braucht die Muskulatur viel Glucose für ihren akut gesteigerten
Stoffwechsel (aktive Skelettmuskeln nehmen Glucose unabhängig vom
Insulinspiegel auf). Adipozyten würden mit der Muskulatur um Glucose
konkurrierten, hoher Sympathikustonus verhindert das, gleichzeitig
fördert er die Glucosefreisetzung in der Leber.
Insulin
regt Zellen, die über Insulinrezeptoren verfügen (primäre Ziele: Adipozyten,
Muskelzellen, Leberzellen) zur Glucoseaufnahme an und kann so den
Blutzuckerspiegel senken. GLUT4-Permeasen sind insulinabhängig (zu Glucosepermeasen s. dort). Epithelzellen in Nieren (Tubuli) und Darm (Mucosa) sowie Nervenzellen exprimieren keine Insulinrezeptoren und sind nicht
insulinabhängig. Andernfalls könnten sie im Hungerzustand keine Glucose
aufnehmen; ihre Glucoseaufnahme ist auch im postresorptiven Zustand gewährleistet (sie verfügen über nicht-insulinabhängige Permeasen für Glucose wie GLUT2, GLUT3 u.a. In Gefahren- bzw. Belastungssituationen wird die Insulinausschüttung gehemmt (Näheres s. dort). Bei solchen fight-or-flight-Situationen
braucht die Muskulatur viel Glucose für ihren akut gesteigerten
Stoffwechsel (aktive Skelettmuskeln nehmen Glucose unabhängig vom
Insulinspiegel auf). Adipozyten würden mit der Muskulatur um Glucose
konkurrierten, hoher Sympathikustonus verhindert das, gleichzeitig
fördert er die Glucosefreisetzung in der Leber. ), gekennzeichnet durch erhöhten Blutzuckerspiegel
(Hyperglykämie ). ) kommen. Da der Zucker Wasser aus osmotischen Gründen "mitnimmt" (osmotische Diurese), kommt es dabei zu vermehrtem Wasserverlust (Diabetes = "Durchfluss"). Als Insulinresistenz bezeichnet man eine verringerte zelluläre Antwort (insulinabhängiger, d.h. mit Insulinrezeptoren ausgestatteter Gewebe) auf Insulin (sowohl körpereigenes als auch exogenes). Dabei ist die Glucosetoleranz
erniedrigt, d.h. Aufnahme einer definierten Glucosemenge führt zu
überhöhtem Anstieg des Blutzuckerspiegels (oGTT: Oraler
Glucosetoleranztest).Insulinresistenz ist ein Element des metabolischen Syndroms ("Syndrom X": Insulinresistenz, Bluthochdruck, Hypertriglyzeridämie, erniedrigtes HDL-Cholesterin, abdominelle Fettleibigkeit) und gehört zu den dringlichsten Gesundheitsproblemen der Wohlstandsgesellschaft.
), gekennzeichnet durch erhöhten Blutzuckerspiegel
(Hyperglykämie ). ) kommen. Da der Zucker Wasser aus osmotischen Gründen "mitnimmt" (osmotische Diurese), kommt es dabei zu vermehrtem Wasserverlust (Diabetes = "Durchfluss"). Als Insulinresistenz bezeichnet man eine verringerte zelluläre Antwort (insulinabhängiger, d.h. mit Insulinrezeptoren ausgestatteter Gewebe) auf Insulin (sowohl körpereigenes als auch exogenes). Dabei ist die Glucosetoleranz
erniedrigt, d.h. Aufnahme einer definierten Glucosemenge führt zu
überhöhtem Anstieg des Blutzuckerspiegels (oGTT: Oraler
Glucosetoleranztest).Insulinresistenz ist ein Element des metabolischen Syndroms ("Syndrom X": Insulinresistenz, Bluthochdruck, Hypertriglyzeridämie, erniedrigtes HDL-Cholesterin, abdominelle Fettleibigkeit) und gehört zu den dringlichsten Gesundheitsproblemen der Wohlstandsgesellschaft.
| Polyurie (osmotische Diurese) ist ein Hauptsymptom eines unbehandelten Diabetes mellitus |
 1869 beschrieb der Pathologe Paul Langerhans in seiner Doktorarbeit die später (1893 durch den Histopathologen Edouard Laguesse)
nach ihm benannnten Zellinseln. Ihre Funktion war zunächst unbekannt. Dass die
Bauchspeicheldrüse mit der Regulierung des Blutzuckerspiegels
zusammenhängt, wurde durch Forschungen von Josef Mering und Oskar Minkowski (um 1900) klar.
Sie konnten zeigen, dass Hunde, denen das Pankreas entfernt wurde,
Diabetes mellitus entwickelten (Minkowski's Labordiener fiel auf, dass
sich am Urin der Tiere Fliegen gütlich taten). Damit bestätigten sie
die Hypothese des französischen Diabetologen Etienne Lanceraux,
dass Diabetes etwas mit einer Fehlfunktion der Bauchspeicheldrüse zu
tun hätte (1877) - eine Hypothese, die im Widerspruch zur Position
stand, die seinerzeit der berühmte Physiologe Claude Bernard vertrat.
1869 beschrieb der Pathologe Paul Langerhans in seiner Doktorarbeit die später (1893 durch den Histopathologen Edouard Laguesse)
nach ihm benannnten Zellinseln. Ihre Funktion war zunächst unbekannt. Dass die
Bauchspeicheldrüse mit der Regulierung des Blutzuckerspiegels
zusammenhängt, wurde durch Forschungen von Josef Mering und Oskar Minkowski (um 1900) klar.
Sie konnten zeigen, dass Hunde, denen das Pankreas entfernt wurde,
Diabetes mellitus entwickelten (Minkowski's Labordiener fiel auf, dass
sich am Urin der Tiere Fliegen gütlich taten). Damit bestätigten sie
die Hypothese des französischen Diabetologen Etienne Lanceraux,
dass Diabetes etwas mit einer Fehlfunktion der Bauchspeicheldrüse zu
tun hätte (1877) - eine Hypothese, die im Widerspruch zur Position
stand, die seinerzeit der berühmte Physiologe Claude Bernard vertrat.  Abbildung).
Abbildung). Abbildung: Insulinempfindliche Gehirnareale und zentrale Insulinwirkungen
Abbildung: Insulinempfindliche Gehirnareale und zentrale Insulinwirkungen Hypothalamus (zentrale metabolische Steuerung), präfrontalen Kortex (Hemmung der Nahrungsaufnahme), Hippocampus
(Gedächtnis und Bewusstseinszustand) und auf den an der Objekterkennung beteiligten gyrus fusiformis
(Identifikation, Belohnung, Emotionslage).
Hypothalamus (zentrale metabolische Steuerung), präfrontalen Kortex (Hemmung der Nahrungsaufnahme), Hippocampus
(Gedächtnis und Bewusstseinszustand) und auf den an der Objekterkennung beteiligten gyrus fusiformis
(Identifikation, Belohnung, Emotionslage).
 s. dort).
s. dort).  Abbildung: Typische Organisation einer Langerhans-Insel in der Bauchspeicheldrüse vgl. dort).
Die Langerhans-Inseln sind stark vaskularisiert, ihre spezifische
Durchblutung beträgt ein Mehrfaches derjeniger des Herzmuskels.
Der Blutstrom zwischen den Inselzellen ist vom Zentrum der jeweiligen
Insel zur Peripherie ausgerichtet (Abbildung), die kapillären
Spalträume zwischen den Inselzellen erfüllen eine pfortaderähnliche
Funktion: So gelangen z.B. Produkte zentraler ß-Zellen relativ
"unverdünnt" zu peripher liegenden F-Zellen.
Abbildung: Typische Organisation einer Langerhans-Insel in der Bauchspeicheldrüse vgl. dort).
Die Langerhans-Inseln sind stark vaskularisiert, ihre spezifische
Durchblutung beträgt ein Mehrfaches derjeniger des Herzmuskels.
Der Blutstrom zwischen den Inselzellen ist vom Zentrum der jeweiligen
Insel zur Peripherie ausgerichtet (Abbildung), die kapillären
Spalträume zwischen den Inselzellen erfüllen eine pfortaderähnliche
Funktion: So gelangen z.B. Produkte zentraler ß-Zellen relativ
"unverdünnt" zu peripher liegenden F-Zellen. Das primäre Translationsprodukt der ß-Zellen ist Präproinsulin, dieses enthält das für den Durchtritt durch die Membran des endoplasmatischen Retikulums erforderliche 24-Aminosäuren-Signalpeptid. Das Signalpeptid wird beim Eintritt in das endoplasmatische Retikulum durch mikrosomale Enzyme abgespalten, wodurch Proinsulin entsteht. Dieses besteht aus drei Teilen: Der A-Kette des Insulins (21 Aminosäuren), dem C-Peptid (31 Aminosäuren; C: connecting)
und der B-Kette des Insulins (30 Aminosäuren). Proinsulin hat ~7% der
biologischen Wirksamkeit des Insulins, ein wenig davon entgeht der
Spaltung zu Insulin und wird zusammen mit diesem von der ß-Zelle
sezerniert. Abbildung).
Das primäre Translationsprodukt der ß-Zellen ist Präproinsulin, dieses enthält das für den Durchtritt durch die Membran des endoplasmatischen Retikulums erforderliche 24-Aminosäuren-Signalpeptid. Das Signalpeptid wird beim Eintritt in das endoplasmatische Retikulum durch mikrosomale Enzyme abgespalten, wodurch Proinsulin entsteht. Dieses besteht aus drei Teilen: Der A-Kette des Insulins (21 Aminosäuren), dem C-Peptid (31 Aminosäuren; C: connecting)
und der B-Kette des Insulins (30 Aminosäuren). Proinsulin hat ~7% der
biologischen Wirksamkeit des Insulins, ein wenig davon entgeht der
Spaltung zu Insulin und wird zusammen mit diesem von der ß-Zelle
sezerniert. Abbildung). Insulin wird vorwiegend in Leber, Nieren und Muskulatur endozytiert (vielleicht ist das für die Hormonwirkung von Bedeutung) und lysosomal abgebaut (IDE, insulin degrading enzyme); seine
Halbwertszeit beträgt etwa 5 Minuten: Kaum aus dem Pankreas in den
Pfortaderkreislauf gelangt, werden ~70% des
neugebildeten Insulins von der Leber abgebaut (first passage),
bevor es den systemischen Kreislauf erreichen kann (das dämpft
Schwankungen des Insulinspiegels im systemischen Kreislauf und bedeutet
gleichzeitig, dass die Leber wesentlich höheren Insulinkonzentrationen
ausgesetzt ist als der Rest des Organismus). In der Niere wird Insulin glomerulär filtriert (nur 51 Aminosäuren!), resorbiert und tubulär zerstückelt. Und auch Muskelzellen lassen Insulinmoleküle nicht weit kommen.) und Amylin
(das vermutlich ähnlich wirkt). Inkretine sind
Verdauungshormone, deren Ausschüttung durch Nahrungsaufnahme angeregt
wird, ihrerseits die Insulinsekretion stimulieren und so eine
zusätzliche Abnahme des Blutzuckerspiegels bewirken. Dieser Inkretineffekt macht
25-60% der gesamten Insulinantwort aus. Er erklärt, warum oral
aufgenommene Glucose stärker insulinanregend wirkt als i.v.
verabreichte. Zu Inkretinen zählen Cholecystokinin, GIP und GLP-1.
Insulin wird vorwiegend in Leber, Nieren und Muskulatur endozytiert (vielleicht ist das für die Hormonwirkung von Bedeutung) und lysosomal abgebaut (IDE, insulin degrading enzyme); seine
Halbwertszeit beträgt etwa 5 Minuten: Kaum aus dem Pankreas in den
Pfortaderkreislauf gelangt, werden ~70% des
neugebildeten Insulins von der Leber abgebaut (first passage),
bevor es den systemischen Kreislauf erreichen kann (das dämpft
Schwankungen des Insulinspiegels im systemischen Kreislauf und bedeutet
gleichzeitig, dass die Leber wesentlich höheren Insulinkonzentrationen
ausgesetzt ist als der Rest des Organismus). In der Niere wird Insulin glomerulär filtriert (nur 51 Aminosäuren!), resorbiert und tubulär zerstückelt. Und auch Muskelzellen lassen Insulinmoleküle nicht weit kommen.) und Amylin
(das vermutlich ähnlich wirkt). Inkretine sind
Verdauungshormone, deren Ausschüttung durch Nahrungsaufnahme angeregt
wird, ihrerseits die Insulinsekretion stimulieren und so eine
zusätzliche Abnahme des Blutzuckerspiegels bewirken. Dieser Inkretineffekt macht
25-60% der gesamten Insulinantwort aus. Er erklärt, warum oral
aufgenommene Glucose stärker insulinanregend wirkt als i.v.
verabreichte. Zu Inkretinen zählen Cholecystokinin, GIP und GLP-1.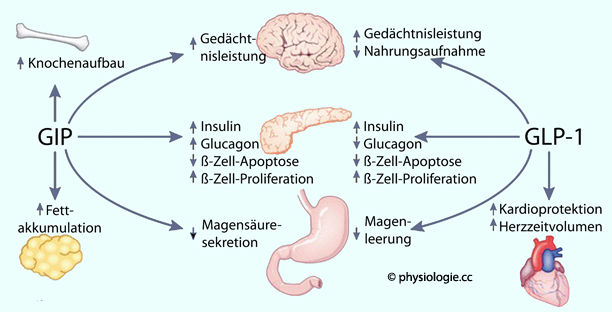 Abbildung: Wirkungen von GIP und GLP-1 Abbildung zeigt, wirken sowohl GIP als auch GLP-1 auf das Gehirn gedächtnisstärkend (GLP-1 steigert auch den Appetit). Im Pankreas
fördern beide die Insulinausschüttung (Inkretineffekt!) sowie
Betazellproliferation und senken die ß-Zell-Apoptoserate; auf die
Glucagonsekretion wirken sie gegenläufig. Am Magen wirkt GIP sekretagog, GLP-1 fördert die Magenentleerung.
Abbildung: Wirkungen von GIP und GLP-1 Abbildung zeigt, wirken sowohl GIP als auch GLP-1 auf das Gehirn gedächtnisstärkend (GLP-1 steigert auch den Appetit). Im Pankreas
fördern beide die Insulinausschüttung (Inkretineffekt!) sowie
Betazellproliferation und senken die ß-Zell-Apoptoserate; auf die
Glucagonsekretion wirken sie gegenläufig. Am Magen wirkt GIP sekretagog, GLP-1 fördert die Magenentleerung. Ein Insulinspiegel von ~120 pM hemmt die Glucoseproduktion halbmaximal, die Glucoseverwertung
ist halbmaximal stimuliert bei ~300 pM.
Ein Insulinspiegel von ~120 pM hemmt die Glucoseproduktion halbmaximal, die Glucoseverwertung
ist halbmaximal stimuliert bei ~300 pM.  s. dort s. dort
s. dort s. dort Inselzellen beeinflussen einander regulatorisch; so hemmt Amylin
die Glucagonsekretion, GLP-1 fördert die Bildung von Insulin und hemmt
die von Glucagon ( s. auch dort). Die Betazellen liegen zentral und modulieren (parakrin, d.h. auf Nachbarzellen) die Aktivität umliegender α-, δ- und PP-Zellen. Sekretionsverhalten:
Bei Werten unter 3 mM extrazellulärer Glucosekonzentration
sezernieren ß-Zellen nur eine basale Mindestmenge Insulin. Bei 5 mM ist
die Bildungsrate bereits verdoppelt und steigt S-förmig mit zunehmendem
Zuckerspiegel an, bis sie bei ~15 mM (etwa dem Vierfachen des normalen Ruhewertes) ein Maximum (beim 4-fachen der Basissekretion) erreicht.
Jeden Tag wird etwa ein
Fünftel des in den Inselzellen gespeicherten Insulins freigesetzt. Die
Insulinkonzentration im Blutplasma kann von der (effektiven) an den
Zielzellen (also im Interstitium) sehr verschieden sein. Die endotheliale Barriere behindert den Übertritt des Insulins ins Zielgewebe. So
kann es bei einer kontinuierlichen Insulininfusion über eine Stunde dauern,
bis der Blutspiegel auch im Interstitium erreicht ist. Der Zeitverlauf
der Glucoseaufnahme (etwa im Muskel) entspricht dem der interstitiellen
Insulinkonzentration, nicht dem des Blut-Insulinspiegels.
Inselzellen beeinflussen einander regulatorisch; so hemmt Amylin
die Glucagonsekretion, GLP-1 fördert die Bildung von Insulin und hemmt
die von Glucagon ( s. auch dort). Die Betazellen liegen zentral und modulieren (parakrin, d.h. auf Nachbarzellen) die Aktivität umliegender α-, δ- und PP-Zellen. Sekretionsverhalten:
Bei Werten unter 3 mM extrazellulärer Glucosekonzentration
sezernieren ß-Zellen nur eine basale Mindestmenge Insulin. Bei 5 mM ist
die Bildungsrate bereits verdoppelt und steigt S-förmig mit zunehmendem
Zuckerspiegel an, bis sie bei ~15 mM (etwa dem Vierfachen des normalen Ruhewertes) ein Maximum (beim 4-fachen der Basissekretion) erreicht.
Jeden Tag wird etwa ein
Fünftel des in den Inselzellen gespeicherten Insulins freigesetzt. Die
Insulinkonzentration im Blutplasma kann von der (effektiven) an den
Zielzellen (also im Interstitium) sehr verschieden sein. Die endotheliale Barriere behindert den Übertritt des Insulins ins Zielgewebe. So
kann es bei einer kontinuierlichen Insulininfusion über eine Stunde dauern,
bis der Blutspiegel auch im Interstitium erreicht ist. Der Zeitverlauf
der Glucoseaufnahme (etwa im Muskel) entspricht dem der interstitiellen
Insulinkonzentration, nicht dem des Blut-Insulinspiegels. Abbildung: Mechanismus der Insulinfreisetzung aus Inselzellen
Second-messenger-Wege s. dort
Abbildung: Mechanismus der Insulinfreisetzung aus Inselzellen
Second-messenger-Wege s. dort Glucose wird (beim Menschen) vorwiegend über einen Glut-1-Transporter in die Zelle aufgenommen. Glut-1 hat einen niedrigeren KM-Wert als Glut-2, dem wichtigsten Glucosetransporter in ß-Zellen von Mäusen (Abbildung) -
dies könnte erklären, warum ß-Zellen des Menschen die Insulinsekretion
schon bei niedrigeren Zuckerspiegeln starten als ß-Zellen bei Mäusen
Glucose wird glykolytisch abgebaut, dies regt die ATP-Synthese an - die Aktivität der Glucokinase ist der limitierende Schritt. Im Gegensatz zu anderen Hexokinasen (z.B. im Skelettmuskel) hat die Glucokinase einen hohen KM-Wert
(12 mM) und durch ihr Produkt Glucose-6-Phosphat nicht inhibiert.
Zusammen mit GLUT2 mit dessen hoher Kapazität dient die Glucokinase als Glucosesensor.
Das System reagiert automatisch: Steigt [Glucose] außerhalb des
Hepatozyten über den Nüchternwert, wird Glucose aufgenommen und
phosphoryliert (dabei reichert sich Glucose-6-Phosphat nicht in der Zelle an, sondern wird zu Glykogen umgebaut, zu Pyruvat glykolysiert, oder in den Pentosephosphatweg eingeschleust)
Anstieg der intrazellulären ATP-Konzentration blockiert ATP-sensible Kaliumkanäle (KATP-Kanäle) - und damit den K+-Ausstrom aus der Zelle (Abbildung),
die Membran depolarisiert (pharmakologische Blockade dieses Kanals -
wie durch Sulfonylharnstoffe - erhöht die Insulinsekretion und senkt
damit den Blutzuckerspiegel)
Depolarisation führt zu Calciumeinstrom - hauptsächlich durch L-Typ-Ca++-Kanäle -, [Ca++]i steigt an, was wiederum
die
Exozytose des Hormons aus Speichervesikeln anregt - allerdings nur
unter Anwesenheit verstärkender Faktoren (wie Citrat bzw.
Membranderivaten wie DAG, 12-S-HETE - die Arachidonsäureproduktion ist ATP-abhängig und damit an den Energiestatus der Zelle geknüpft).
Glucose wird (beim Menschen) vorwiegend über einen Glut-1-Transporter in die Zelle aufgenommen. Glut-1 hat einen niedrigeren KM-Wert als Glut-2, dem wichtigsten Glucosetransporter in ß-Zellen von Mäusen (Abbildung) -
dies könnte erklären, warum ß-Zellen des Menschen die Insulinsekretion
schon bei niedrigeren Zuckerspiegeln starten als ß-Zellen bei Mäusen
Glucose wird glykolytisch abgebaut, dies regt die ATP-Synthese an - die Aktivität der Glucokinase ist der limitierende Schritt. Im Gegensatz zu anderen Hexokinasen (z.B. im Skelettmuskel) hat die Glucokinase einen hohen KM-Wert
(12 mM) und durch ihr Produkt Glucose-6-Phosphat nicht inhibiert.
Zusammen mit GLUT2 mit dessen hoher Kapazität dient die Glucokinase als Glucosesensor.
Das System reagiert automatisch: Steigt [Glucose] außerhalb des
Hepatozyten über den Nüchternwert, wird Glucose aufgenommen und
phosphoryliert (dabei reichert sich Glucose-6-Phosphat nicht in der Zelle an, sondern wird zu Glykogen umgebaut, zu Pyruvat glykolysiert, oder in den Pentosephosphatweg eingeschleust)
Anstieg der intrazellulären ATP-Konzentration blockiert ATP-sensible Kaliumkanäle (KATP-Kanäle) - und damit den K+-Ausstrom aus der Zelle (Abbildung),
die Membran depolarisiert (pharmakologische Blockade dieses Kanals -
wie durch Sulfonylharnstoffe - erhöht die Insulinsekretion und senkt
damit den Blutzuckerspiegel)
Depolarisation führt zu Calciumeinstrom - hauptsächlich durch L-Typ-Ca++-Kanäle -, [Ca++]i steigt an, was wiederum
die
Exozytose des Hormons aus Speichervesikeln anregt - allerdings nur
unter Anwesenheit verstärkender Faktoren (wie Citrat bzw.
Membranderivaten wie DAG, 12-S-HETE - die Arachidonsäureproduktion ist ATP-abhängig und damit an den Energiestatus der Zelle geknüpft). | Wirkungskette Insulin: Glucoseeinstrom über GLUT2 → Glykolyse, ATP-Synthese → Blockade ATP-sensitiven K+-Ausstroms → Depolarisation → Ca++-Einstrom über L-Typ-Calciumkanäle → [Ca++]i steigt → Exozytose, Insulinfreisetzung |

 Abbildung: Funktion der Betazelle abhängig vom Glucosespiegel
Abbildung: Funktion der Betazelle abhängig vom Glucosespiegel teils verstärkt (Glucagon, GIP, GLP-1, ß2-adrenerg, cholinerg),
teils verstärkt (Glucagon, GIP, GLP-1, ß2-adrenerg, cholinerg),  teils abgeschwächt (Somatostatin, α2-adrenerg).
Abbildung unten):
teils abgeschwächt (Somatostatin, α2-adrenerg).
Abbildung unten): Zunächst durch Exozytose aus bereits nahe der Membran der Betazellen gelegenen, fusionsbereiten Vesikeln (readily releasable pool)
Dann durch Rekrutierung tiefer gelegener Vesikel ("Speichergranula"; weniger intensive, aber anhaltende Sekretion) sowie Neusynthese.
Die Freisetzung von Insulin aus dem Pankreas wird physiologisch angeregt durch
Zunächst durch Exozytose aus bereits nahe der Membran der Betazellen gelegenen, fusionsbereiten Vesikeln (readily releasable pool)
Dann durch Rekrutierung tiefer gelegener Vesikel ("Speichergranula"; weniger intensive, aber anhaltende Sekretion) sowie Neusynthese.
Die Freisetzung von Insulin aus dem Pankreas wird physiologisch angeregt durch Aktivität des Parasympathikus (cholinerg über M-Rezeptoren) sowie des Sympathikus (ß2-adrenerg) Anstieg des Blutzuckerspiegels (Wirkungskette s. Abbildung: "Glucosespiegel hoch") Anstieg der Konzentration einiger Amino- (Alanin, Arginin, verzweigtkettige) und Fettsäuren Freigesetztes
Insulin (autokrines positives Feedback, Selbstverstärkung der
Insulinsekretion). Dass Insulin pulsatil freigesetzt wird, verhindert
vermutlich eine Desensibilisierung (durch receptor downregulation) Wirkung einiger gastrointestinaler Hormone (vor allem GIP; GLP-1, Abbildung; auch CCK, Gastrin, Sekretin). Diese bewirken über Steigerung der cAMP-Konzentration in der ß-Zelle den Inkretineffekt, d.h. orale Zufuhr von Glucose wirkt sich stärker auf die Insulinfreisetzung aus als eine intravenöse Gabe derselben Dosis Osteocalcin
Aktivität des Parasympathikus (cholinerg über M-Rezeptoren) sowie des Sympathikus (ß2-adrenerg) Anstieg des Blutzuckerspiegels (Wirkungskette s. Abbildung: "Glucosespiegel hoch") Anstieg der Konzentration einiger Amino- (Alanin, Arginin, verzweigtkettige) und Fettsäuren Freigesetztes
Insulin (autokrines positives Feedback, Selbstverstärkung der
Insulinsekretion). Dass Insulin pulsatil freigesetzt wird, verhindert
vermutlich eine Desensibilisierung (durch receptor downregulation) Wirkung einiger gastrointestinaler Hormone (vor allem GIP; GLP-1, Abbildung; auch CCK, Gastrin, Sekretin). Diese bewirken über Steigerung der cAMP-Konzentration in der ß-Zelle den Inkretineffekt, d.h. orale Zufuhr von Glucose wirkt sich stärker auf die Insulinfreisetzung aus als eine intravenöse Gabe derselben Dosis Osteocalcin Abbildung: Wie GLP-1 in der Betazelle die Insulinsekretion anregt
Abbildung: Wie GLP-1 in der Betazelle die Insulinsekretion anregt Epac, exchange protein associated with cAMP, Proteine, die MAP-Kinasen aktivieren ER, endoplasmatisches Retikulum Pdx-1, pancreatic and duodenal homeobox 1, Transkriptionsfaktor mit fördernder Wirkung auf Pankreas und Dünndarm PKA, Proteinkinase A
RYR, Ryanodin-Calciumkanal VDCC, spannungsbetriebener Calciumkanal
Epac, exchange protein associated with cAMP, Proteine, die MAP-Kinasen aktivieren ER, endoplasmatisches Retikulum Pdx-1, pancreatic and duodenal homeobox 1, Transkriptionsfaktor mit fördernder Wirkung auf Pankreas und Dünndarm PKA, Proteinkinase A
RYR, Ryanodin-Calciumkanal VDCC, spannungsbetriebener Calciumkanal Inkretine aus der Dünndarmschleimhaut
"melden", dass Nährstoffe resorbiert werden und veranlassen die
ß-Zellen des Pankreas zu verstärkter Antwort auf die Glucosebelastung (feed-forward-Effekt).
Inkretine aus der Dünndarmschleimhaut
"melden", dass Nährstoffe resorbiert werden und veranlassen die
ß-Zellen des Pankreas zu verstärkter Antwort auf die Glucosebelastung (feed-forward-Effekt). Inkretin-Analoga eignen
sich gut zur insulinabhängigen Blutzuckersenkung bei Diabetikern: Sie
federn die Gefahr einer Hypoglykämie ab, denn wenn der Glucosespiegel sinkt, hört ihre Wirkung auf die Insulinfreisetzung auf. Die Freisetzung von Insulin aus dem Pankreas wird physiologisch gehemmt durch Sympathikusaktivität (über α2-Rezeptoren, welche [cAMP] senken; ß2-Rezeptoren stimulieren gleichzeitig die Glykogenolyse / Gluconeogenese in
Muskulatur und Leber, beides wirkt
blutzuckersteigernd). Bei körperlicher Arbeit wird so die Insulinausschüttung gesenkt, und so kommt es bei Belastung
nicht nur zu Senkung des Blutzuckerspiegels (vermehrter Verbrauch durch
die arbeitende Muskulatur), sondern auch des Insulinspiegels (bei gut
Trainierten bis auf die Hälfte des Ruhewertes) Weiters hemmen mehrere
Peptide die Insulinsekretion, wie
Somatostatin aus den D-Zellen (dessen Freisetzungs u.a. durch Adrenalin angeregt wird) der Cotransmitter
Galanin Amylin - auch Insel-Amyloid-Polypeptid (IAPP) genannt -, das zusammen mit Insulin aus der ß-Zelle stammt und vermutlich durch Hemmung
der Glucagonsekretion den Blutzuckerspiegel stabilisiert Auch Leptin (aus Fettgewebe) wirkt sich auf Insulin aus: Es hemmt - über eine Leptinrezeptor-assoziierte Januskinase
- sowohl die Transkription und Biosynthese des Insulins als auch (über
Öffnung von Kaliumkanälen) dessen Freisetzung aus der Betazelle. Je
mehr das Fettgewebe zunimmt, umso stärker wirkt sich der hemmende
Effekt des Leptins auf die Insulinsekretion aus.
Inkretin-Analoga eignen
sich gut zur insulinabhängigen Blutzuckersenkung bei Diabetikern: Sie
federn die Gefahr einer Hypoglykämie ab, denn wenn der Glucosespiegel sinkt, hört ihre Wirkung auf die Insulinfreisetzung auf. Die Freisetzung von Insulin aus dem Pankreas wird physiologisch gehemmt durch Sympathikusaktivität (über α2-Rezeptoren, welche [cAMP] senken; ß2-Rezeptoren stimulieren gleichzeitig die Glykogenolyse / Gluconeogenese in
Muskulatur und Leber, beides wirkt
blutzuckersteigernd). Bei körperlicher Arbeit wird so die Insulinausschüttung gesenkt, und so kommt es bei Belastung
nicht nur zu Senkung des Blutzuckerspiegels (vermehrter Verbrauch durch
die arbeitende Muskulatur), sondern auch des Insulinspiegels (bei gut
Trainierten bis auf die Hälfte des Ruhewertes) Weiters hemmen mehrere
Peptide die Insulinsekretion, wie
Somatostatin aus den D-Zellen (dessen Freisetzungs u.a. durch Adrenalin angeregt wird) der Cotransmitter
Galanin Amylin - auch Insel-Amyloid-Polypeptid (IAPP) genannt -, das zusammen mit Insulin aus der ß-Zelle stammt und vermutlich durch Hemmung
der Glucagonsekretion den Blutzuckerspiegel stabilisiert Auch Leptin (aus Fettgewebe) wirkt sich auf Insulin aus: Es hemmt - über eine Leptinrezeptor-assoziierte Januskinase
- sowohl die Transkription und Biosynthese des Insulins als auch (über
Öffnung von Kaliumkanälen) dessen Freisetzung aus der Betazelle. Je
mehr das Fettgewebe zunimmt, umso stärker wirkt sich der hemmende
Effekt des Leptins auf die Insulinsekretion aus.| Die Insulinsekretion wird u.a. gehemmt durch sympathische Aktivität (via α-Rezeptoren) und Somatostatin (aus D-Zellen) |
Abbildung):  Abbildung: Phasenweise Freisetzung von Insulin ins Blut In der zephalen
Phase erklärt sich die vagale Stimulation durch die Wahrnehmung der
Nahrung (z.B. wenn das Essen auf den Tisch kommt - Anblick, Geruch). Diese präabsorptive
Phase dauert etwa 10 Minuten. Sie ist unabhängig von der Freisetzung
von GIP oder GLP1, sie wird hauptsächlich vagal (parasympathisch)
mediiert; in der gastrischen Phase durch Einflüsse aus dem Magen, insbesondere Gastrinfreisetzung; in der intestinalen
Phase durch das Anströmen von Substratmolekülen ("Substratphase" -
Anstieg des Glucosespiegels!). Letztere hält am längsten an und gibt
einen intensiven Effekt auf die Insulinfreisetzung.
Abbildung: Phasenweise Freisetzung von Insulin ins Blut In der zephalen
Phase erklärt sich die vagale Stimulation durch die Wahrnehmung der
Nahrung (z.B. wenn das Essen auf den Tisch kommt - Anblick, Geruch). Diese präabsorptive
Phase dauert etwa 10 Minuten. Sie ist unabhängig von der Freisetzung
von GIP oder GLP1, sie wird hauptsächlich vagal (parasympathisch)
mediiert; in der gastrischen Phase durch Einflüsse aus dem Magen, insbesondere Gastrinfreisetzung; in der intestinalen
Phase durch das Anströmen von Substratmolekülen ("Substratphase" -
Anstieg des Glucosespiegels!). Letztere hält am längsten an und gibt
einen intensiven Effekt auf die Insulinfreisetzung. Abbildung: Rückkopplungsschleifen der Blutzuckerregulation s. dort) kennzeichnet den Status des Energiestoffwechsels ( Abbildung oben):
Abbildung: Rückkopplungsschleifen der Blutzuckerregulation s. dort) kennzeichnet den Status des Energiestoffwechsels ( Abbildung oben): Es ist hoch nach dem Essen
(Resorptionsphase; viel Insulin), im Überschuss vorhandene Glucose
wird
gespeichert In der Postresorptionsphase ist es niedrig (wenig Insulin),
die Energiespeicher werden angegriffen.
Es ist hoch nach dem Essen
(Resorptionsphase; viel Insulin), im Überschuss vorhandene Glucose
wird
gespeichert In der Postresorptionsphase ist es niedrig (wenig Insulin),
die Energiespeicher werden angegriffen. Einfluss des Stoffwechselstatus Nach Boron / Boulpaep: Concise Medical Physiology, Elsevier 2021 |
||
| Variable / Organ |
Nach 24 Stunden Nahrungskarenz |
2 Stunden nach Nahrungsaufnahme (Mischkost) |
| Blutzuckerspiegel |
60-80 mg/dl 3,3-4,4 mM |
100-140 mg/dl 5,6-7,8 mM |
| Insulinspiegel |
3-8 µU/ml |
50-150 µU/ml |
| Glucagonspiegel |
40-80 pg/ml |
80-200 pg/ml |
| Leber |
↑Glykogenolyse ↑Gluconeogenese |
↑Glykogensynthese ↓Glykogenolyse ↓Gluconeogenese |
| Fettgewebe |
Mobilisierung von Lipiden |
Synthese von Lipiden |
| Muskelgewebe |
Metabolisierung von Lipiden Proteinabbau, Amibosäurenexport |
Glucose oxidiert oder als Glykogen gespeichert Proteine gespart |
Abbildung):  Ras-abhängig über IRS1 / IRS2 und das Adapterprotein Grb-2 (growth factor receptor-bound protein 2) und Aktivierung der RAS / MAPK Kaskade und Einfluss auf die Gentranskription (z.B. für Glucokinase) im Zellkern. Die Aktivierungsschritte sind die folgenden: Bindung des Insulinmoleküls an den Rezeptor (liegt als Dimer vor) Autophosphorylierung der Tyrosinreste am Rezeptor Über die Phosphotyrosinreste binden Dockingproteine wie IRS-1 Der Insulinrezeptor phosphoryliert IRS-1 (IRS-2 bindet an andere Adapterproteine, hat ähnliche Funktion) An das phosphorylierte IRS-1 binden Adapterproteine (Grb2) Diese Bindung leitet die Aktivierung von Ras und der MAPK-Kaskade ein... ...Proteine im Zellkern, welche die Transkription von Glucokinase steigern, werden phosphoryliert Ras-unabhängig über IRS1, Aktivierung von Phosphoinositid-3-Kinasen (PI3K) und Proteinkinase B
und nachfolgende Proteinphosphorylierungen, was z.B. Aktivierung der
Glykogensynthase oder die Einlagerung von Glucosetransportern (GLUT4) -
und damit Glucoseaufnahme in die Zelle - zur Folge hat. Bindung des Insulinmoleküls an den Rezeptor Autophosphorylierung der Tyrosinreste am Rezeptor Über die Phosphotyrosinreste binden Dockingproteine wie IRS-1 Der Insulinrezeptor phosphoryliert IRS-1 (bis zu diesem Schritt wie bein Ras-abhängigen Weg) Phosphoryliertes IRS-1 aktiviert PI3-Kinase, die Phosphatidylinositolphosphate (PIP2, PIP3) generiert
Diese membrangebundenen Phosphoinositide wirken als second messengers,
schalten Proteinkinase B (PKB) ein, die durch Phosphorylierung
aktiviert wird
PKB ändert die Aktivität zahlreicher Proteine in der Zelle, die nun die
Aufnahme und Speicherung von Glucose fördern und die Glykogensynthese
anregen
Ras-abhängig über IRS1 / IRS2 und das Adapterprotein Grb-2 (growth factor receptor-bound protein 2) und Aktivierung der RAS / MAPK Kaskade und Einfluss auf die Gentranskription (z.B. für Glucokinase) im Zellkern. Die Aktivierungsschritte sind die folgenden: Bindung des Insulinmoleküls an den Rezeptor (liegt als Dimer vor) Autophosphorylierung der Tyrosinreste am Rezeptor Über die Phosphotyrosinreste binden Dockingproteine wie IRS-1 Der Insulinrezeptor phosphoryliert IRS-1 (IRS-2 bindet an andere Adapterproteine, hat ähnliche Funktion) An das phosphorylierte IRS-1 binden Adapterproteine (Grb2) Diese Bindung leitet die Aktivierung von Ras und der MAPK-Kaskade ein... ...Proteine im Zellkern, welche die Transkription von Glucokinase steigern, werden phosphoryliert Ras-unabhängig über IRS1, Aktivierung von Phosphoinositid-3-Kinasen (PI3K) und Proteinkinase B
und nachfolgende Proteinphosphorylierungen, was z.B. Aktivierung der
Glykogensynthase oder die Einlagerung von Glucosetransportern (GLUT4) -
und damit Glucoseaufnahme in die Zelle - zur Folge hat. Bindung des Insulinmoleküls an den Rezeptor Autophosphorylierung der Tyrosinreste am Rezeptor Über die Phosphotyrosinreste binden Dockingproteine wie IRS-1 Der Insulinrezeptor phosphoryliert IRS-1 (bis zu diesem Schritt wie bein Ras-abhängigen Weg) Phosphoryliertes IRS-1 aktiviert PI3-Kinase, die Phosphatidylinositolphosphate (PIP2, PIP3) generiert
Diese membrangebundenen Phosphoinositide wirken als second messengers,
schalten Proteinkinase B (PKB) ein, die durch Phosphorylierung
aktiviert wird
PKB ändert die Aktivität zahlreicher Proteine in der Zelle, die nun die
Aufnahme und Speicherung von Glucose fördern und die Glykogensynthese
anregen Abbildung: RAS-abhängige und -unabhängige Wege für insulinstimulierte intrazelluläre Signalkaskaden Abbildungen ). Bindet Insulin an seinen Rezeptor, autophosphoryliert dieser, das Phosphotyrosin wird vom Dockingprotein IRS (insulin receptor substrate) erkannt und gebunden. Dieses akiviert in weiterer Folge Phosphatidylinositol-3-Kinase (PI3K); resultierendes PIP2 / PIP3
schaltet über eine weitere Kinase (PDK) Proteinkinase B ein, und diese
bewirkt die Mobilisierung (aus intrazellulären Depots) und Insertion
von Glucosetransportmolekülen (GLUT4) in die äußere Membran der Zelle,
die nun Glucose aus dem Interstitium aufnehmen kann. Als Insulinempfindlichkeit bezeichnet man das Ausmaß an insulinabhängiger Glucoseaufnahme in die Zelle.
Abbildung: RAS-abhängige und -unabhängige Wege für insulinstimulierte intrazelluläre Signalkaskaden Abbildungen ). Bindet Insulin an seinen Rezeptor, autophosphoryliert dieser, das Phosphotyrosin wird vom Dockingprotein IRS (insulin receptor substrate) erkannt und gebunden. Dieses akiviert in weiterer Folge Phosphatidylinositol-3-Kinase (PI3K); resultierendes PIP2 / PIP3
schaltet über eine weitere Kinase (PDK) Proteinkinase B ein, und diese
bewirkt die Mobilisierung (aus intrazellulären Depots) und Insertion
von Glucosetransportmolekülen (GLUT4) in die äußere Membran der Zelle,
die nun Glucose aus dem Interstitium aufnehmen kann. Als Insulinempfindlichkeit bezeichnet man das Ausmaß an insulinabhängiger Glucoseaufnahme in die Zelle. Abbildung: Signaltransduktion nach Aktivierung des Insulinrezeptors FOXO1, forkhead box protein O1, ein Transkriptionsfaktor, der im Insulin-Signalweg Gluconeogenese und Glykolyse reguliert G6Pase, Glucose-6-Phosphatase, hydrolysiert Glucose-6-Phosphat GRB2, Growth-factor receptor-bound protein 2, Adapterprotein GS, Glykogensynthase GSK, GS-Kinase IF, Initiation factor, Proteinkomplexe, helfen bei der mRNA-Translation IRS, insulin receptor substrate, Adapterproteine JNK, Proteinkinase MAPK, Mitogenaktivierte Proteinkinase MEK, Mitogenaktivierte Proteinkinase-Kinase mTOR, target of rapamycin, eine Kinase p38, Proteinkinase PDK, Phosphoinositide- dependent kinase, eine "Master"-Kinase in der Insulin- Signalkette PEPCK, Phosphoenolpyruvate carboxykinase, gluconeogenetische Lyase PHAS-1, phosphorylated heat- and acid- stable protein, Initiation der Translation regulierendes Protein PI3K, Phosphatidylinositol 3-Kinase Raf-1, nach rapidly accelerated fibrosarcoma, Proteinkinase Ras (nach rat sarcoma), eine GTPase SH2, SRC homology domain 2, Interaktion von Proteinen vermittelnde Proteindomäne SHC, src homology domain C terminus, ein Transformationsprotein SOS, son of sevenless, Guaninnukleotid- AustauschfaktorAbbildung).
Abbildung: Signaltransduktion nach Aktivierung des Insulinrezeptors FOXO1, forkhead box protein O1, ein Transkriptionsfaktor, der im Insulin-Signalweg Gluconeogenese und Glykolyse reguliert G6Pase, Glucose-6-Phosphatase, hydrolysiert Glucose-6-Phosphat GRB2, Growth-factor receptor-bound protein 2, Adapterprotein GS, Glykogensynthase GSK, GS-Kinase IF, Initiation factor, Proteinkomplexe, helfen bei der mRNA-Translation IRS, insulin receptor substrate, Adapterproteine JNK, Proteinkinase MAPK, Mitogenaktivierte Proteinkinase MEK, Mitogenaktivierte Proteinkinase-Kinase mTOR, target of rapamycin, eine Kinase p38, Proteinkinase PDK, Phosphoinositide- dependent kinase, eine "Master"-Kinase in der Insulin- Signalkette PEPCK, Phosphoenolpyruvate carboxykinase, gluconeogenetische Lyase PHAS-1, phosphorylated heat- and acid- stable protein, Initiation der Translation regulierendes Protein PI3K, Phosphatidylinositol 3-Kinase Raf-1, nach rapidly accelerated fibrosarcoma, Proteinkinase Ras (nach rat sarcoma), eine GTPase SH2, SRC homology domain 2, Interaktion von Proteinen vermittelnde Proteindomäne SHC, src homology domain C terminus, ein Transformationsprotein SOS, son of sevenless, Guaninnukleotid- AustauschfaktorAbbildung).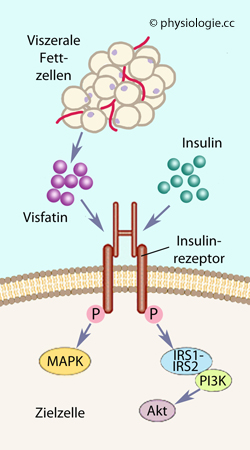 Abbildung: Anregung des Insulinrezeptors IRS, Insulinrezeptor-Substrat
- vermittelt Insulinwirkung auf intrazelluläre Pfade wie PI3K =
Phosphoinositid-3-Kinase und Akt = (Gene der) Proteinkinase B Über Glucosetransporter (GLUT) s. dort Abbildung) ist ein Proteohormon, das in viszeralem Fettgewebe (=im freien Bauchraum um die inneren Organe angelagertes Fett, auch intraabdominales Fett) gebildet wird. Sein Plasmaspiegel korreliert mit dem Grad einer
Adipositas. Zu seinen Effekten zählen weiters die Bildung und Speicherung von Glykogen (Anregung der Glykogensynthase in Leber und
Muskel) die Proteinsynthese (Aminosäureaufnahme) die Entfernung (Clearance) von Chylomikronen aus dem Blut (s. auch dort) Triglyzeridsynthese und Fetteinlagerung Glucoseaufnahme (Einbau von GLUT 4 in Fett- und Muskelzellen) und Glucoseverbrauch in Leber-, Muskel- und Fettzellen Aufnahme
von Kalium, Calcium,
Nukleosiden, Phosphat (Hyperkaliämie lässt sich mit i.v.-Insulin-Glucose-Gabe behandeln) - die Kaliumaufnahme wird durch Anregung der Na/K-ATPase intensiviert; Wachstum und Genexpression. Langfristig wirkt Insulin (vor allem während der
fetalen Entwicklung) wachstumsfördernd (über Insulinrezeptoren s. auch dort). den Proteinabbau in peripheren Geweben die Glucoseabgabe der Leber die Produktion von VLDL in der Leber die Lipolyse: Insulin ist das einzige
Hormon, das - über eine hormonsensitive Lipase - die Lipolyse hemmt und so die Fettdepots schützt die Aktivität der Fruktose-1,6-Biphosphatase, des Schlüsselenzyms der
Gluconeogenese (in einer Situation des Glucoseüberflusses nicht
gefragt) die Ausscheidung von Phosphat
Abbildung: Anregung des Insulinrezeptors IRS, Insulinrezeptor-Substrat
- vermittelt Insulinwirkung auf intrazelluläre Pfade wie PI3K =
Phosphoinositid-3-Kinase und Akt = (Gene der) Proteinkinase B Über Glucosetransporter (GLUT) s. dort Abbildung) ist ein Proteohormon, das in viszeralem Fettgewebe (=im freien Bauchraum um die inneren Organe angelagertes Fett, auch intraabdominales Fett) gebildet wird. Sein Plasmaspiegel korreliert mit dem Grad einer
Adipositas. Zu seinen Effekten zählen weiters die Bildung und Speicherung von Glykogen (Anregung der Glykogensynthase in Leber und
Muskel) die Proteinsynthese (Aminosäureaufnahme) die Entfernung (Clearance) von Chylomikronen aus dem Blut (s. auch dort) Triglyzeridsynthese und Fetteinlagerung Glucoseaufnahme (Einbau von GLUT 4 in Fett- und Muskelzellen) und Glucoseverbrauch in Leber-, Muskel- und Fettzellen Aufnahme
von Kalium, Calcium,
Nukleosiden, Phosphat (Hyperkaliämie lässt sich mit i.v.-Insulin-Glucose-Gabe behandeln) - die Kaliumaufnahme wird durch Anregung der Na/K-ATPase intensiviert; Wachstum und Genexpression. Langfristig wirkt Insulin (vor allem während der
fetalen Entwicklung) wachstumsfördernd (über Insulinrezeptoren s. auch dort). den Proteinabbau in peripheren Geweben die Glucoseabgabe der Leber die Produktion von VLDL in der Leber die Lipolyse: Insulin ist das einzige
Hormon, das - über eine hormonsensitive Lipase - die Lipolyse hemmt und so die Fettdepots schützt die Aktivität der Fruktose-1,6-Biphosphatase, des Schlüsselenzyms der
Gluconeogenese (in einer Situation des Glucoseüberflusses nicht
gefragt) die Ausscheidung von PhosphatInsulineffekte auf Kohlenhydrat-, Fett- und Proteinstoffwechsel Nach Ritter / Flower / Henderson / Loke / MacEwan / Rang, Rang & Dale's Pharmacology, 9th ed. Elsevier 2020 |
|||
| Metabolismus |
Hepatozyten |
Adipozyten |
Myozyten |
| Kohlenhydrate |
↓Gluconeogenese ↓Glykogenolyse ↑Glykolyse ↑Glykogensynthese |
↑Glucoseaufnahme ↑Glycerinsynthese |
↑Glucoseaufnahme ↑Glykolyse ↑Glykogensynthese |
| Fette |
↑Fettaufbau ↓Lipolyse |
↑Triglyzeridsynthese ↑Fettsäuresynthese ↓Lipolyse |
- |
| Proteine |
↓Proteinabbau | - |
↑Aminosäurenaufnahme ↑Proteinsynthese |
Abbildung).  Abbildung: Insulinwirkungen auf HepatozytenAbbildung) Glykogensynthese und Glykogenolyse:
Insulin begünstigt die Bildung frischen Glykogens und hemmt dessen
Abbau. Glucose gelangt via GLUT2 (insulin-unabhängig) in die Zelle.Glucokinase und Glykogensynthase
(dessen Dephosphorylierung seine Aktivität steigert) werden durch
Insulin angeregtsowohl Glucose als auch Insulin senken die Aktivität
der Glykogenphosphorylase (was den Glykogenaufbau fördert)Insulin hemmt die Glucose-6-Phosphatase, was die Konvertierung von G6P zu Glucose reduziert.
Glykolyse und Gluconeogenese: Insulin begünstigt auch den Abbau von
Glucose zu Pyruvat, das über mitochondrielles Acetyl-Coenzym A zur
Bildung von Triglyzeriden genutzt wird. Insulin fördert die Transkription des Glucokinase-Gens; dascurch wird mehr Glucose zu G6P phosphoryliert.Insulin stimuliert Phosphofructokinase-1 (PFK-1), das geschwindigkeitsbestimmende Enzym der Glykolyse. PFK-1 fördert die Phosphorylierung von Fructose-6-Phosphat zu Fructose 1,6-Biphosphat. PFK-1wird aktiviert durch AMP und Fructose-2,6-Biphosphat (dessen Spiegel durch Insulin ansteigt), gehemmt durch ATP und Citrat. Insulin regt (Phosphatasen) die Aktivität der Pyruvatkinase (diese bildet irreversibel Pyruvat aus Phosphoenolpyruvat) und der Pyruvatdehydrogenase (die Pyruvat oxidiert) an. Lipogenese: Die Triglyzeridsynthese wird angeregt, und Malonyl-CoA hemmt mitochondrielle Carnitin-Acyltransferase 1
(CAT 1) - und damit den Fettsäuretransport in die Mitochondrien (wo
Fettsäuren oxidiert würden). CAT 1 bildet Acylcarnitin, was notwendig
ist, damit langkettige Fettsäuren die innere Mitochondrienmembran
passieren können. Insulin sorgt für Dephosphorylierung der Acetyl-Coenzym A Carboxylase 2
(ACC2), was wiederum - über vermehrt entstehendes Malonyl-CoA - zu
allosterischer Hemmung der CAT 1 führt. Insulin aktiviert gleichzeitig Fettsäuresynthase und damit die Bildung von Neutralfetten. Proteinmetabolismus: Über komplexe Wege wird die Synthese von Eiweiß angeregt und sein Abbau gehemmt. Abbildung), Aminosäuretransportern und
Na-K-ATPase in die Plasmamembran, was u.a. die Eiweißsynthese und
Aufnahme
von Kaliumionen erleichtert.
Abbildung: Insulinwirkungen auf HepatozytenAbbildung) Glykogensynthese und Glykogenolyse:
Insulin begünstigt die Bildung frischen Glykogens und hemmt dessen
Abbau. Glucose gelangt via GLUT2 (insulin-unabhängig) in die Zelle.Glucokinase und Glykogensynthase
(dessen Dephosphorylierung seine Aktivität steigert) werden durch
Insulin angeregtsowohl Glucose als auch Insulin senken die Aktivität
der Glykogenphosphorylase (was den Glykogenaufbau fördert)Insulin hemmt die Glucose-6-Phosphatase, was die Konvertierung von G6P zu Glucose reduziert.
Glykolyse und Gluconeogenese: Insulin begünstigt auch den Abbau von
Glucose zu Pyruvat, das über mitochondrielles Acetyl-Coenzym A zur
Bildung von Triglyzeriden genutzt wird. Insulin fördert die Transkription des Glucokinase-Gens; dascurch wird mehr Glucose zu G6P phosphoryliert.Insulin stimuliert Phosphofructokinase-1 (PFK-1), das geschwindigkeitsbestimmende Enzym der Glykolyse. PFK-1 fördert die Phosphorylierung von Fructose-6-Phosphat zu Fructose 1,6-Biphosphat. PFK-1wird aktiviert durch AMP und Fructose-2,6-Biphosphat (dessen Spiegel durch Insulin ansteigt), gehemmt durch ATP und Citrat. Insulin regt (Phosphatasen) die Aktivität der Pyruvatkinase (diese bildet irreversibel Pyruvat aus Phosphoenolpyruvat) und der Pyruvatdehydrogenase (die Pyruvat oxidiert) an. Lipogenese: Die Triglyzeridsynthese wird angeregt, und Malonyl-CoA hemmt mitochondrielle Carnitin-Acyltransferase 1
(CAT 1) - und damit den Fettsäuretransport in die Mitochondrien (wo
Fettsäuren oxidiert würden). CAT 1 bildet Acylcarnitin, was notwendig
ist, damit langkettige Fettsäuren die innere Mitochondrienmembran
passieren können. Insulin sorgt für Dephosphorylierung der Acetyl-Coenzym A Carboxylase 2
(ACC2), was wiederum - über vermehrt entstehendes Malonyl-CoA - zu
allosterischer Hemmung der CAT 1 führt. Insulin aktiviert gleichzeitig Fettsäuresynthase und damit die Bildung von Neutralfetten. Proteinmetabolismus: Über komplexe Wege wird die Synthese von Eiweiß angeregt und sein Abbau gehemmt. Abbildung), Aminosäuretransportern und
Na-K-ATPase in die Plasmamembran, was u.a. die Eiweißsynthese und
Aufnahme
von Kaliumionen erleichtert. 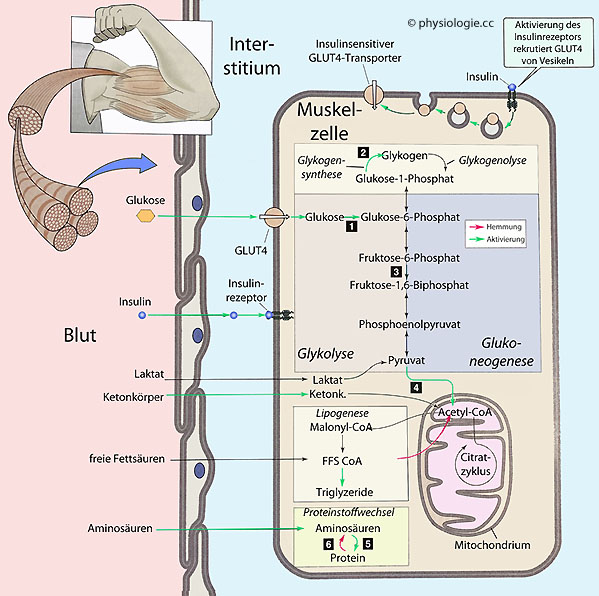 Abbildung: Insulinwirkungen auf Myozyten
Abbildung: Insulinwirkungen auf Myozyten| Insulin
fördert den Einbau von GLUT-4 in Skelettmuskelzellen, diese nehmen
dadurch vermehrt Glucose auf - der Blutzuckerspiegel sinkt |
| Insulin fördert die K+-Aufnahme in den Muskel durch Anregung der Na/K-ATPase Insulininjektion kann zu akuter Hypokaliämie führen |
| Insulin regt die Proteinsynthese in Skelettmuskelzellen an |
 Abbildung: Insulinwirkungen auf Adipozyten Förderung der Glucoseaufnahme durch Einlagerung von (insulinsensitivem) GLUT4 aus intrazellulären Speichern in die Zellmembran Anregung der Glykolyse sowie der Umwandlung von Pyruvat (Pyruvat-Dehydrogenase (1) und Acetyl-CoA-Carboxylase (2) werden durch Insulin angeregt) zu metabolischen Speicherformen (vor allem Fettsäuren, kaum Glykogen) Triglyzeridsynthese und Speicherung in Fetttröpfchen. Die Aktivität der Triglyzeridlipase und der hormonsensitiven Lipase (Box 3) wird durch Insulin reduziert (diese würden Triglyzeride zu Glycerin und freien Fettsäuren abbauen) Synthese von Lipoproteinlipase, die von Fett- an Endothelzellen exportiert
und an deren Blutseite verankert wird (Abspaltung von Triglyzeriden aus Chylomikronen und VLDL - die
Fettsäuren gelangen zu den Adipozyten, diese bauen sie in ihren
Triglyzeridpool ein)
Abbildung: Insulinwirkungen auf Adipozyten Förderung der Glucoseaufnahme durch Einlagerung von (insulinsensitivem) GLUT4 aus intrazellulären Speichern in die Zellmembran Anregung der Glykolyse sowie der Umwandlung von Pyruvat (Pyruvat-Dehydrogenase (1) und Acetyl-CoA-Carboxylase (2) werden durch Insulin angeregt) zu metabolischen Speicherformen (vor allem Fettsäuren, kaum Glykogen) Triglyzeridsynthese und Speicherung in Fetttröpfchen. Die Aktivität der Triglyzeridlipase und der hormonsensitiven Lipase (Box 3) wird durch Insulin reduziert (diese würden Triglyzeride zu Glycerin und freien Fettsäuren abbauen) Synthese von Lipoproteinlipase, die von Fett- an Endothelzellen exportiert
und an deren Blutseite verankert wird (Abspaltung von Triglyzeriden aus Chylomikronen und VLDL - die
Fettsäuren gelangen zu den Adipozyten, diese bauen sie in ihren
Triglyzeridpool ein)
| Insulin hemmt die Lipolyse |
mangelnde Hormonbildung (insuffiziente B-Zellen im Pankreas - Typ-1-Diabetes) Insulinunempfindlichkeit der Peripherie (blockierte bzw.
fehlende Insulinrezeptoren - Typ-2-Diabetes).  Abbildung: Insulininjektion in das Unterhautfettgewebe
Abbildung: Insulininjektion in das Unterhautfettgewebe| Metabolische
Azidose (Ketoazidose) mit vertiefter Atmung (respiratorische
Kompensation) ist typisch für unbehandelten Diabetes mellitus |
Zu den schweren gesundheitlichen Konsequenzen eines nicht oder unzureichend behandelten Diabetes mellitus gehören Nierenversagen, Neuropathien, Infarkte, Erblindung. 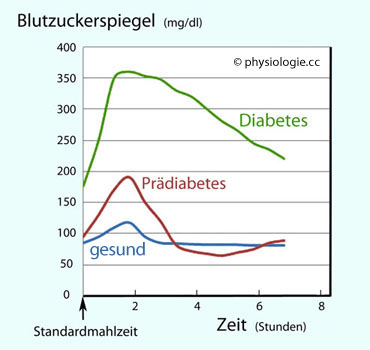 Abbildung: Oraler Glucose-ToleranztestAbbildung).
Da das Insulin eine kurze Halbwertszeit (~5 min) hat, sagt seine
Serumkonzentration wenig über die längerfristige Regulation aus. Der Anteil des HbA1C am gesamten Hämoglobin im Blut
liegt bei Nichtdiabetikern unter 5%
(<50 mM HbA1C /M Hb). Bei Werten über 9% (>90 mM/M) werden die mit
Diabetes mellitus einhergehenden Gesundheitsrisiken als extrem erhöht eingestuft. Sulfonylharnstoffe
hemmen den ATP-abhängigen Kaliumkanal der Betazellen (s.
oben) und erhöhen dadurch die Insulinfreisetzung (Therapie bei Diabetes
Typ II).
Abbildung: Oraler Glucose-ToleranztestAbbildung).
Da das Insulin eine kurze Halbwertszeit (~5 min) hat, sagt seine
Serumkonzentration wenig über die längerfristige Regulation aus. Der Anteil des HbA1C am gesamten Hämoglobin im Blut
liegt bei Nichtdiabetikern unter 5%
(<50 mM HbA1C /M Hb). Bei Werten über 9% (>90 mM/M) werden die mit
Diabetes mellitus einhergehenden Gesundheitsrisiken als extrem erhöht eingestuft. Sulfonylharnstoffe
hemmen den ATP-abhängigen Kaliumkanal der Betazellen (s.
oben) und erhöhen dadurch die Insulinfreisetzung (Therapie bei Diabetes
Typ II).| Sulfonylharnstoffe
senken den Blutzuckerspiegel durch Blockade des ATP-sensitiven
Kaliumkanals der pankreatischen Betazelle und Insulinfreisetzung |
 Fall 1
Fall 1
 Zunahme des Glucosespiegels um ~50% verdreifacht den Insulinspiegel. Insulin fördert durch Einlagerung insulinabhängiger Glucosetransporter (GLUT4) in
die Zellmembran Glucoseaufnahme und Energiespeicherung
vor allem in Fett- und Muskelgewebe, der Blutzuckerspiegel sinkt. Insulin wirkt auf die Funktionen des Gehirns, Anstieg des Insulinspiegels ist auch ein Sättigungssignal
Proinsulin besteht aus der A-Kette des Insulins, dem C-Peptid und der
B-Kette des Insulins. Insulin wird zusammen mit abgespaltenem C-Peptid
in sekretorischen Granula der ß-Zellen in Form von Zink-Komplexen
gespeichert. Der Insulinvorrat der Bauchspeicheldrüse beträgt etwa 10
mg (250 IE), jeden Tag wird etwa ein Fünftel des in den Inselzellen
gespeicherten Insulins freigesetzt. Die
basale Insulinfreisetzung beträgt beim Erwachsenen ~1 IE/h. ß-Zellen
liegen im Inneren der Inseln, freigesetztes Insulin diffundiert zu
äußeren
Inselzellen und moduliert die Aktivität von α-, δ- und PP-Zellen.
ß-Zellen
sind über gap
junctions synchronisiert, Insulin wird alle 3-6 Minuten freigesetzt.
Der Blutspiegel kann zwischen <40 pM (nüchtern) und ~1000 pM
schwanken (postprandial). Die Insulinkonzentration im Blut und im
Interstitium verhalten sich unterschiedlich: Die endotheliale Barriere
behindert den Übertritt in das Zielgewebe, wo der Zeitverlauf der Glucoseaufnahme dem der
interstitiellen, nicht dem der Insulinkonzentration im Blut entspricht.
Insulin wird in Leber (~50%), Nieren und Muskulatur
lysosomal abgebaut, seine Halbwertszeit beträgt ~5 Minuten Aufnahme von Glucose, Galaktose, Mannose oder Aminosäuren (Arginin,
Leuzin u.a.) regt den Stoffwechsel der ß-Zelle an. Das steigert den
ATP-Gehalt, senkt den Kalium-Ausstrom (ATP-sensitiver Kaliumkanal),
führt zu Depolarisierung, Calciumeinstrom und Insulinfreisetzung. Das
mitausgeschiedene C-Peptid ist im Blut ein
Indikator der endogenen Insulinproduktion (injizierte Insulinpräparate
entalten kein C-Peptid). Die glucoseabhängige Insulinfreisetzung wird
u.a. verstärkt durch Glucagon, GIP, GLP-1, cholinerg;
abgeschwächt α2-adrenerg und durch Somatostatin Die Insulinfreisetzung wird entsprechend den Verdauungsphasen
reguliert: In der zephalen Phase parasympathisch, in der gastrischen
Phase durch Gastrin, in der intestinalen Phase durch Substratmoleküle (längste Dauer, intensiver Effekt). Das
Konzentrationsverhältnis Insulin / Glucagon kennzeichnet den Status des
Energiestoffwechsels (Resorptionsphase viel Insulin,
Postresorptionsphase wenig Insulin). Insulin fördert die Aufnahme,
Verwertung und Speicherung von Glucose, Lipiden und Aminosäuren,
Glykogenbildung, Lipogenese und Proteinsynthese. Als Insulinempfindlichkeit
bezeichnet man das Ausmaß an insulinabhängiger Glucoseaufnahme in die
Zellen. Aktivierung des Insulinrezeptors (einer Tyrosinkinase) hat
metabolische und wachstumsfördernde Effekte. Zielproteine in der Zelle
werden phosphoryliert und intrazelluläre Signalwege aktiviert. Der
Insulinrezeptor wird rasch internalisiert (Refrakterität)
In Fettzellen hemmt Insulin die Lipolyse und regt die Aufnahme von
Glucose sowie die Speicherung von Fettsäuren an. In Muskelzellen
stimuliert es die Einlagerung von Aminosäuretransportern und
Na/K-ATPase in die Zellmembran. Hexokinasen phosphorylieren Glucose zu
Glucose-6-Phosphat, das in der Zelle "gefangen" ist und über
Isomerisierung zu Glucose-1-Phosphat zu Glykogen polymerisiert werden
kann, oder in die Glykolyse bzw. den Pentosephosphatweg geleitet wird.
Die Insulinwirkungen auf den Kohlenhydrat- und Fettstoffwechsel sowie
auf den Transport von Aminosäuren und Kaliumionen erfolgt im Sekunden-
bis Minutenbereich. Auf die Leber wirkt Insulin innerhalb von ~20
Minuten nach Beginn der Nahrungsaufnahme (Gluconeogenese), die Anregung
der Glucoseaufnahme in der Peripherie beginnt nach etwa einer Stunde.
Zellwachstum wird erst nach Tagen merklich angeregt
Metabolische Azidose (Ketoazidose) mit vertiefter Atmung
(respiratorische Kompensation) ist typisch für unbehandelten Diabetes
mellitus. Sulfonylharnstoffe senken den Blutzuckerspiegel durch
Blockade des ATP-sensitiven Kaliumkanals der Betazelle, was die
Insulinfreisetzung anregt Zunahme des Glucosespiegels um ~50% verdreifacht den Insulinspiegel. Insulin fördert durch Einlagerung insulinabhängiger Glucosetransporter (GLUT4) in
die Zellmembran Glucoseaufnahme und Energiespeicherung
vor allem in Fett- und Muskelgewebe, der Blutzuckerspiegel sinkt. Insulin wirkt auf die Funktionen des Gehirns, Anstieg des Insulinspiegels ist auch ein Sättigungssignal
Proinsulin besteht aus der A-Kette des Insulins, dem C-Peptid und der
B-Kette des Insulins. Insulin wird zusammen mit abgespaltenem C-Peptid
in sekretorischen Granula der ß-Zellen in Form von Zink-Komplexen
gespeichert. Der Insulinvorrat der Bauchspeicheldrüse beträgt etwa 10
mg (250 IE), jeden Tag wird etwa ein Fünftel des in den Inselzellen
gespeicherten Insulins freigesetzt. Die
basale Insulinfreisetzung beträgt beim Erwachsenen ~1 IE/h. ß-Zellen
liegen im Inneren der Inseln, freigesetztes Insulin diffundiert zu
äußeren
Inselzellen und moduliert die Aktivität von α-, δ- und PP-Zellen.
ß-Zellen
sind über gap
junctions synchronisiert, Insulin wird alle 3-6 Minuten freigesetzt.
Der Blutspiegel kann zwischen <40 pM (nüchtern) und ~1000 pM
schwanken (postprandial). Die Insulinkonzentration im Blut und im
Interstitium verhalten sich unterschiedlich: Die endotheliale Barriere
behindert den Übertritt in das Zielgewebe, wo der Zeitverlauf der Glucoseaufnahme dem der
interstitiellen, nicht dem der Insulinkonzentration im Blut entspricht.
Insulin wird in Leber (~50%), Nieren und Muskulatur
lysosomal abgebaut, seine Halbwertszeit beträgt ~5 Minuten Aufnahme von Glucose, Galaktose, Mannose oder Aminosäuren (Arginin,
Leuzin u.a.) regt den Stoffwechsel der ß-Zelle an. Das steigert den
ATP-Gehalt, senkt den Kalium-Ausstrom (ATP-sensitiver Kaliumkanal),
führt zu Depolarisierung, Calciumeinstrom und Insulinfreisetzung. Das
mitausgeschiedene C-Peptid ist im Blut ein
Indikator der endogenen Insulinproduktion (injizierte Insulinpräparate
entalten kein C-Peptid). Die glucoseabhängige Insulinfreisetzung wird
u.a. verstärkt durch Glucagon, GIP, GLP-1, cholinerg;
abgeschwächt α2-adrenerg und durch Somatostatin Die Insulinfreisetzung wird entsprechend den Verdauungsphasen
reguliert: In der zephalen Phase parasympathisch, in der gastrischen
Phase durch Gastrin, in der intestinalen Phase durch Substratmoleküle (längste Dauer, intensiver Effekt). Das
Konzentrationsverhältnis Insulin / Glucagon kennzeichnet den Status des
Energiestoffwechsels (Resorptionsphase viel Insulin,
Postresorptionsphase wenig Insulin). Insulin fördert die Aufnahme,
Verwertung und Speicherung von Glucose, Lipiden und Aminosäuren,
Glykogenbildung, Lipogenese und Proteinsynthese. Als Insulinempfindlichkeit
bezeichnet man das Ausmaß an insulinabhängiger Glucoseaufnahme in die
Zellen. Aktivierung des Insulinrezeptors (einer Tyrosinkinase) hat
metabolische und wachstumsfördernde Effekte. Zielproteine in der Zelle
werden phosphoryliert und intrazelluläre Signalwege aktiviert. Der
Insulinrezeptor wird rasch internalisiert (Refrakterität)
In Fettzellen hemmt Insulin die Lipolyse und regt die Aufnahme von
Glucose sowie die Speicherung von Fettsäuren an. In Muskelzellen
stimuliert es die Einlagerung von Aminosäuretransportern und
Na/K-ATPase in die Zellmembran. Hexokinasen phosphorylieren Glucose zu
Glucose-6-Phosphat, das in der Zelle "gefangen" ist und über
Isomerisierung zu Glucose-1-Phosphat zu Glykogen polymerisiert werden
kann, oder in die Glykolyse bzw. den Pentosephosphatweg geleitet wird.
Die Insulinwirkungen auf den Kohlenhydrat- und Fettstoffwechsel sowie
auf den Transport von Aminosäuren und Kaliumionen erfolgt im Sekunden-
bis Minutenbereich. Auf die Leber wirkt Insulin innerhalb von ~20
Minuten nach Beginn der Nahrungsaufnahme (Gluconeogenese), die Anregung
der Glucoseaufnahme in der Peripherie beginnt nach etwa einer Stunde.
Zellwachstum wird erst nach Tagen merklich angeregt
Metabolische Azidose (Ketoazidose) mit vertiefter Atmung
(respiratorische Kompensation) ist typisch für unbehandelten Diabetes
mellitus. Sulfonylharnstoffe senken den Blutzuckerspiegel durch
Blockade des ATP-sensitiven Kaliumkanals der Betazelle, was die
Insulinfreisetzung anregt |