

Eine Reise durch die Physiologie - Wie der Körper des Menschen funktioniert


 Leber und
Fettstoffwechsel
Leber und
Fettstoffwechsel
 Carnitin: caro, carnis (lat) = Fleisch
Carnitin: caro, carnis (lat) = Fleisch| Im Lipidmetabolismus spielt die Leber mehrere Rollen: -- Sie baut Fettsäuren ab (Beta-Oxidation), um Energie zu mobilisieren; Hepatozyten "exportieren" dabei Acetessigsäure, die in Empfängerzellen über Acetyl-Coenzym A weiterverwertet wird (Citratzyklus) -- Sie bildet aus Kohlenhydraten körpereigene Neutralfette, Phosphoglyzeride, Sphingolipide, Cholesterin, Gallensäuren, Lezithin -- Sie bildet Apoproteine für den Lipidtransport sowie VLDL-Lipoproteine -- Sie synthetisiert Cholesterin, das als Bestandteil der Zellmembranen und für den Aufbau von Steroidhormonen und Gallensäuren benötigt wird. Fettlösliche Vitamine werden zu einem beträchtlichen Anteil in der Leber (vor allem den Ito-Zellen) gespeichert. Chronische Lebererkrankungen äußern sich u.a. in reduziertem Lipoproteinspiegel im Blut. |
 Sortierung absorbierter Lipide Hepatische Cholesterinproduktion Abbau von Fettsäuren Hungerstoffwechsel Gallensalze Leber & Vitamine
Sortierung absorbierter Lipide Hepatische Cholesterinproduktion Abbau von Fettsäuren Hungerstoffwechsel Gallensalze Leber & Vitamine
 Core messages
Core messages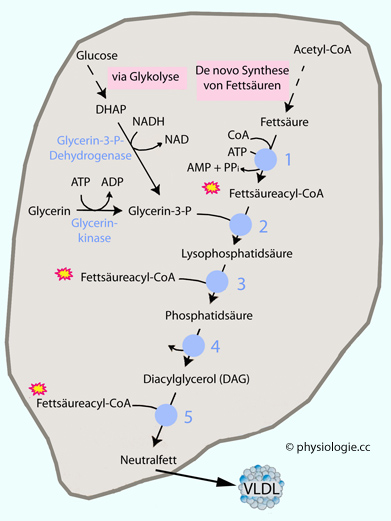
 Abbildung: Triglyceridsynthese in Hepatozyten
Abbildung: Triglyceridsynthese in Hepatozyten Abbildung). Diese werden dann von den Leberzellen in VLDL-Partikel eingebaut und in dieser Form an die Blutbahn abgegeben.
Abbildung). Diese werden dann von den Leberzellen in VLDL-Partikel eingebaut und in dieser Form an die Blutbahn abgegeben.  sind
sind  Energiespeicher (Neutralfette),
Bestandteile von Membranen (z.B. Phospholipide, Cholesterin ), Steroidhormone (Geschlechtshormone, Corticoide, Vit-D3-Hormon) und Gallensäuren (Cholsäure,
Chenodesoxycholsäure, ..).
Energiespeicher (Neutralfette),
Bestandteile von Membranen (z.B. Phospholipide, Cholesterin ), Steroidhormone (Geschlechtshormone, Corticoide, Vit-D3-Hormon) und Gallensäuren (Cholsäure,
Chenodesoxycholsäure, ..).
 Abbildung: Stoffwechsel der Fettsäuren in der Leber
Abbildung: Stoffwechsel der Fettsäuren in der Leber Abbildung: Leber und Lipidkreislauf
Abbildung: Leber und Lipidkreislauf IDL = intermediate density lipoprotein
LDL
= low density lipoprotein
LPL
= Lipoproteinlipase, an Endothelzellen gebundenes Enzym, spaltet Triglyceride aus Lipoproteinen ab
VLDL = very low density lipoproteinAbbildung). Dies geschieht vor
allem in der postresorptiven
(Hunger-) Phase, in der Substrate für den Energiestoffwechsel nicht
mehr aus dem Darm kommen und von der Leber beigestellt werden.
IDL = intermediate density lipoprotein
LDL
= low density lipoprotein
LPL
= Lipoproteinlipase, an Endothelzellen gebundenes Enzym, spaltet Triglyceride aus Lipoproteinen ab
VLDL = very low density lipoproteinAbbildung). Dies geschieht vor
allem in der postresorptiven
(Hunger-) Phase, in der Substrate für den Energiestoffwechsel nicht
mehr aus dem Darm kommen und von der Leber beigestellt werden. Der Triglyzeridnachschub für die Peripherie erfolgt in der resorptiven Phase vorwiegend über Chylomikronen, in der postresorptiven Phase über VLDL.
Der Triglyzeridnachschub für die Peripherie erfolgt in der resorptiven Phase vorwiegend über Chylomikronen, in der postresorptiven Phase über VLDL. Synposis der Lipoproteinfunktionen s. dort
Synposis der Lipoproteinfunktionen s. dort Kurzkettige Fettsäuren
(bis C6) sind relativ gut löslich und werden z.B. direkt über die Pfortader
transportiert. Langkettige Fettsäuren (>C12), Cholesterin u.a. werden hingegen vom
Darm in Chylomikronen "verpackt" und dann in Lymphe und systemischem Kreislauf weiterbefördert.
Kurzkettige Fettsäuren
(bis C6) sind relativ gut löslich und werden z.B. direkt über die Pfortader
transportiert. Langkettige Fettsäuren (>C12), Cholesterin u.a. werden hingegen vom
Darm in Chylomikronen "verpackt" und dann in Lymphe und systemischem Kreislauf weiterbefördert.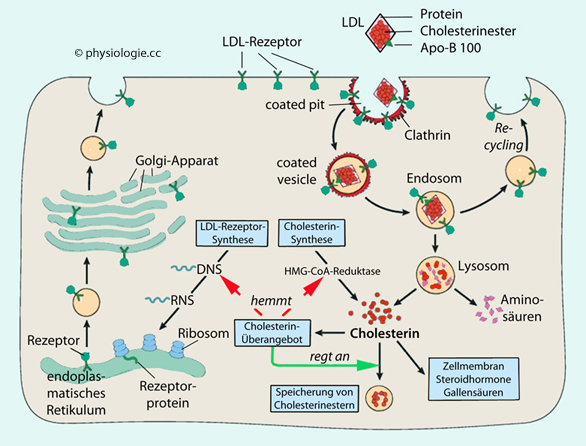 Abbildung: Regulierung des Cholesterinstoffwechsels in einer Leberzelle
Abbildung: Regulierung des Cholesterinstoffwechsels in einer Leberzelle zum Zweck der Energiegewinnung und anderer metabolischer Schritte
Fettsäuren abzubauen (zu oxidieren). Beta-Oxidation erfolgt in Leberzellen besonders
rasch. Essigsäure kann über die Atmungskette vollständig abgebaut werden (zu CO2 und H2O), aus
resorbierten Kohlenhydraten, Aminosäuren und Fetten körpereigene
Lipide (Triglyceride = Neutralfette, Phosphoglyzeride, Sphingolipide, Cholesterin, Gallensäuren, Lezithin) zu
synthetisieren, Apoproteine für den Transport von Triglyceriden im Blut zu synthetisieren, VLDL-Lipoproteine (extrazellulär transportierbares Fett) aufzubauen und in die Blutbahn
zu sezernieren (diese münden in die Lipid-Zirkulation ein und werden von anderen Geweben verwendet).
zum Zweck der Energiegewinnung und anderer metabolischer Schritte
Fettsäuren abzubauen (zu oxidieren). Beta-Oxidation erfolgt in Leberzellen besonders
rasch. Essigsäure kann über die Atmungskette vollständig abgebaut werden (zu CO2 und H2O), aus
resorbierten Kohlenhydraten, Aminosäuren und Fetten körpereigene
Lipide (Triglyceride = Neutralfette, Phosphoglyzeride, Sphingolipide, Cholesterin, Gallensäuren, Lezithin) zu
synthetisieren, Apoproteine für den Transport von Triglyceriden im Blut zu synthetisieren, VLDL-Lipoproteine (extrazellulär transportierbares Fett) aufzubauen und in die Blutbahn
zu sezernieren (diese münden in die Lipid-Zirkulation ein und werden von anderen Geweben verwendet). 

 Abbildung: Cholesterinmetabolismus CE, Cholesterinester FA, Fettsäure(n) LCAT, Lecithin-cholesterol acyltransferase LPL, Lipoproteinlipase SR-B1, scavenger receptor B1 (HDL-Rezeptor) TG, Triglycerid(e)Abbildung oben).
Abbildung: Cholesterinmetabolismus CE, Cholesterinester FA, Fettsäure(n) LCAT, Lecithin-cholesterol acyltransferase LPL, Lipoproteinlipase SR-B1, scavenger receptor B1 (HDL-Rezeptor) TG, Triglycerid(e)Abbildung oben).  Wie entfernt der Körper Cholesterin aus dem Blut? Dieser und anderen damit verbundenen Fragen gingen die Amerikaner Michael Brown und Joseph Goldstein
nach. Unter anderem entdeckten sie die LDL-Rezeptoren auf menschlichen
Zellen und fanden heraus, dass ein Mangel dieser Rezeptoren zu
Hypercholesterinämie führt. "Für ihre Entdeckung zur Bestimmung des
Cholesterinumsatzes" erhielten sie 1985 den Nobelpreis für Physiologie
oder Medizin Cholesterin ist im Blut an Proteine (Apoproteine) gebunden - vorwiegend in Form von LDL- und HDL-Lipoproteinen - und
kann daher nicht direkt in / durch Zellmembranen diffundieren (obwohl
es lipophil ist). Für die Aufnahme in eine Zelle bedarf es der
Wechselwirkung von Lipoproteinen und entsprechenden Rezeptoren
(LDL-Rezeptoren, HDL- ("scavenger") Rezeptoren).
Wie entfernt der Körper Cholesterin aus dem Blut? Dieser und anderen damit verbundenen Fragen gingen die Amerikaner Michael Brown und Joseph Goldstein
nach. Unter anderem entdeckten sie die LDL-Rezeptoren auf menschlichen
Zellen und fanden heraus, dass ein Mangel dieser Rezeptoren zu
Hypercholesterinämie führt. "Für ihre Entdeckung zur Bestimmung des
Cholesterinumsatzes" erhielten sie 1985 den Nobelpreis für Physiologie
oder Medizin Cholesterin ist im Blut an Proteine (Apoproteine) gebunden - vorwiegend in Form von LDL- und HDL-Lipoproteinen - und
kann daher nicht direkt in / durch Zellmembranen diffundieren (obwohl
es lipophil ist). Für die Aufnahme in eine Zelle bedarf es der
Wechselwirkung von Lipoproteinen und entsprechenden Rezeptoren
(LDL-Rezeptoren, HDL- ("scavenger") Rezeptoren). Statine hemmen die HMG-CoA-Reduktase und damit die Cholesterinsynthese im endoplasmatischen Retikulum, sie heißen auch Cholesterin-Syntheseenzym-Hemmer. Sinkt
die hepatische
Cholesterinbildung, wird die Synthese von LDL-Rezeptoren
hinaufreguliert, und die Clearance von LDL aus dem Blut zur Leber
steigt an. Damit senken Statine den Cholesterinspiegel im Blut.
Statine hemmen die HMG-CoA-Reduktase und damit die Cholesterinsynthese im endoplasmatischen Retikulum, sie heißen auch Cholesterin-Syntheseenzym-Hemmer. Sinkt
die hepatische
Cholesterinbildung, wird die Synthese von LDL-Rezeptoren
hinaufreguliert, und die Clearance von LDL aus dem Blut zur Leber
steigt an. Damit senken Statine den Cholesterinspiegel im Blut.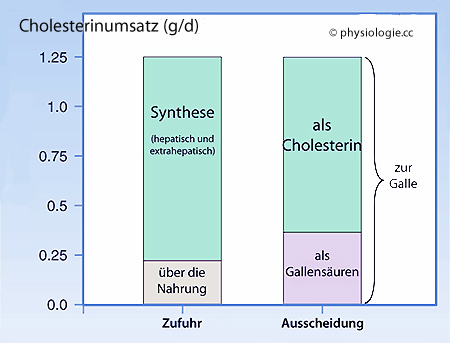 Abbildung: Typische Werte für den täglichen Cholesterinumsatz einer erwachsenen Person (vereinfacht)
Abbildung: Typische Werte für den täglichen Cholesterinumsatz einer erwachsenen Person (vereinfacht) Cholesterin aus der Nahrung
(resorbiert werden bis zu ~0,3 g/d, steigerbar bis ~0,5 g/d) -
normalerweise ~10-20% des Neubedarfs. Die Regulierung der Balance
Eigensynthese - intestinale Aufnahme erfolgt über mehrere Mechanismen,
z.B. Hemmung der HMG-CoA-Reduktase (reduziert
β-Hydroxy-β-Methylglutaryl-Coenzym-A zu Mevalonat) durch Cholesterin
selbst. Bei Bedarf kann die Aktivität
der HMG-CoA-Reduktase bis ~200-fach ansteigen.
Cholesterin aus der Nahrung
(resorbiert werden bis zu ~0,3 g/d, steigerbar bis ~0,5 g/d) -
normalerweise ~10-20% des Neubedarfs. Die Regulierung der Balance
Eigensynthese - intestinale Aufnahme erfolgt über mehrere Mechanismen,
z.B. Hemmung der HMG-CoA-Reduktase (reduziert
β-Hydroxy-β-Methylglutaryl-Coenzym-A zu Mevalonat) durch Cholesterin
selbst. Bei Bedarf kann die Aktivität
der HMG-CoA-Reduktase bis ~200-fach ansteigen.| Cholesterinsynthese ~1 g/d (0,5-2,0) |
Cholesterin aus LDL
(LDL transportiert körpereigenes - hepatisch gebildetes - Cholesterin
an die Gewebe) - Cholesterin bindet an LDL-Rezeptoren und wird so aus
der Blutbahn entfernt. Cholesterin unterliegt körpereigenem Recycling. Neusynthese von 1-2 g/d. Abbildung: Cholesterin in Zellmembranen und im Blut Abbildung). Der hepatische Energiebedarf wird weitgehend durch Fettsäureoxidation (zu Acetyl-Coenzym A) gedeckt. Abbildung):In Phase I gelangen die Fettsäuren (FS) in die Matrix der Mitochondrien. Das geht bei kurzkeittigen FS (SCFAs: short-chain fatty acids) und mittelkettigen (bis 14C: MCFAs, medium-chain) direkt, bei langkettigen (LCFAs: long-chain) und sehr langkettigen (VLCFAs: very long-chain - ab 20C)
bedarf es anderer Mechanismen. LCFAs können aus dem Zytoplasma des
Hepatozyten nur mittels eines sogenannten Carnitin-Shuttle (s. unten)
in die Mitochondrien gebracht werden, VLCFAs werden in Peroxisomen so
lange oxidiert, bis sie LCFA-Länge erreicht haben (<20C).In Phase II erfolgt dann die ß-Oxidation: Die Fettsäuren werdem - je nach Kettenlänge komplett oder teilweise - abgebaut.
Abbildung: Cholesterin in Zellmembranen und im Blut Abbildung). Der hepatische Energiebedarf wird weitgehend durch Fettsäureoxidation (zu Acetyl-Coenzym A) gedeckt. Abbildung):In Phase I gelangen die Fettsäuren (FS) in die Matrix der Mitochondrien. Das geht bei kurzkeittigen FS (SCFAs: short-chain fatty acids) und mittelkettigen (bis 14C: MCFAs, medium-chain) direkt, bei langkettigen (LCFAs: long-chain) und sehr langkettigen (VLCFAs: very long-chain - ab 20C)
bedarf es anderer Mechanismen. LCFAs können aus dem Zytoplasma des
Hepatozyten nur mittels eines sogenannten Carnitin-Shuttle (s. unten)
in die Mitochondrien gebracht werden, VLCFAs werden in Peroxisomen so
lange oxidiert, bis sie LCFA-Länge erreicht haben (<20C).In Phase II erfolgt dann die ß-Oxidation: Die Fettsäuren werdem - je nach Kettenlänge komplett oder teilweise - abgebaut. Abbildung: Fettsäureverwertung in Mitochondrien
Abbildung: Fettsäureverwertung in Mitochondrien Die Kopplung findet auf der zytoplasmatischen Seite der Membran statt,
der Fettsäureacyl-CoA-Komplex passiert die äußere Mitochondrienmembran
und gelangt in den Intermembranraum, wo die Fettsäure von CoA auf die quaternäre Ammoniumverbindung Carnitin (ein Kation) übertragen wird. Dies erfolgt durch Carnitin Palmitoyltransferase
CPT1, das geschwindigkeitslimitierende Enzym der Reaktionskette (die
Reaktion findet an der Innenseite der Membran statt). Carnitin
transportiert langkettige Fettsäuren aus dem Zytoplasma in
Mitochondrien, wo diese oxidiert werden und freie Energie gewonnen
wird. ).
Die Kopplung findet auf der zytoplasmatischen Seite der Membran statt,
der Fettsäureacyl-CoA-Komplex passiert die äußere Mitochondrienmembran
und gelangt in den Intermembranraum, wo die Fettsäure von CoA auf die quaternäre Ammoniumverbindung Carnitin (ein Kation) übertragen wird. Dies erfolgt durch Carnitin Palmitoyltransferase
CPT1, das geschwindigkeitslimitierende Enzym der Reaktionskette (die
Reaktion findet an der Innenseite der Membran statt). Carnitin
transportiert langkettige Fettsäuren aus dem Zytoplasma in
Mitochondrien, wo diese oxidiert werden und freie Energie gewonnen
wird. ).  Abbildung:
Zellulärer Substratmangel führt zu Ketonkörperbildung in der LeberAbbildung) drei Moleküle Acetyl-Coenzym A (AcCoA) zu 3-Hydroxy-3-Methylglutaryl CoA (HMG CoA). Dieses wird zu Acetoacetat (Acetessigsäure) und weiter in ß-Hydroxybuttersäure umgewandelt, oder spontan zu Aceton decarboxyliert ("Ketonkörper").
1,3-Hydroxybutyrat Dehydrogenase oxidiert ß-Hydroxybuttersäure
NAD-abhängig zu Acetessigsäure. NADH spendet dann seine Elektronen an
den Komplex I der Atmungskette und es entstehen 3 Moleküle ATP. In
einem mehrstufigen enzymatischen Prozess wird Acetessigsäure durch
Kopplung mit CoA aktiviert, in 2 AcCoA umgewandelt und in den
Krebszyklus eingeschleust - es entstehen 12 Moleküle ATP. Nicht
benötigtes AcCoA wird durch Kondensation zu Acetessigsäure umgewandelt (nur die Leber kann das), diese kann von
Gehirn, Muskeln und Nieren für deren Energiestoffwechsel verwendet
werden (nicht von der Leber selbst). Zum Hungerstoffwechsel s. auch dort
Abbildung:
Zellulärer Substratmangel führt zu Ketonkörperbildung in der LeberAbbildung) drei Moleküle Acetyl-Coenzym A (AcCoA) zu 3-Hydroxy-3-Methylglutaryl CoA (HMG CoA). Dieses wird zu Acetoacetat (Acetessigsäure) und weiter in ß-Hydroxybuttersäure umgewandelt, oder spontan zu Aceton decarboxyliert ("Ketonkörper").
1,3-Hydroxybutyrat Dehydrogenase oxidiert ß-Hydroxybuttersäure
NAD-abhängig zu Acetessigsäure. NADH spendet dann seine Elektronen an
den Komplex I der Atmungskette und es entstehen 3 Moleküle ATP. In
einem mehrstufigen enzymatischen Prozess wird Acetessigsäure durch
Kopplung mit CoA aktiviert, in 2 AcCoA umgewandelt und in den
Krebszyklus eingeschleust - es entstehen 12 Moleküle ATP. Nicht
benötigtes AcCoA wird durch Kondensation zu Acetessigsäure umgewandelt (nur die Leber kann das), diese kann von
Gehirn, Muskeln und Nieren für deren Energiestoffwechsel verwendet
werden (nicht von der Leber selbst). Zum Hungerstoffwechsel s. auch dort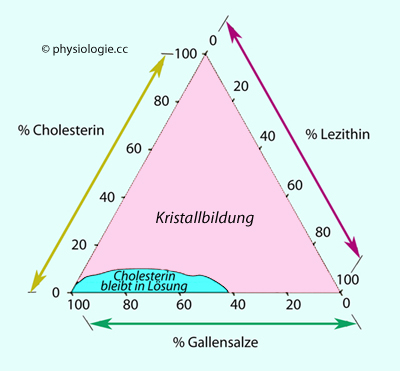 Abbildung: In-vitro-Modell für die Löslichkeit in der Gallenflüssigkeit gelöster potentieller Steinbildner Vitamin A (~80% in Stellatumzellen) Vitamin D (25-Hydroxylierung in der Leber) Vitamin E Vitamin K (>Gerinnung s. dort)Zyanokobalamin hat die längste Halbwertszeit aller im Körper gespeicherten Vitamine: Im Kreislauf ca. 6 Tage, in der Leber ca. ein Jahr. Freie Fettsäuren werden zur Bildung von VLDL herangezogen; bei Unterernährung oder
Diabetes mellitus (Kohlenhydratmangel der Zellen) zur Bildung von Ketonkörpern (Acetessigsäure,
ß-OH-Buttersäure, Aceton). Diese werden von der Leber nicht benötigt,
sondern dienen der Energieabdeckung extrahepatischer Gewebe (Gehirn
u.a.).
Abbildung: In-vitro-Modell für die Löslichkeit in der Gallenflüssigkeit gelöster potentieller Steinbildner Vitamin A (~80% in Stellatumzellen) Vitamin D (25-Hydroxylierung in der Leber) Vitamin E Vitamin K (>Gerinnung s. dort)Zyanokobalamin hat die längste Halbwertszeit aller im Körper gespeicherten Vitamine: Im Kreislauf ca. 6 Tage, in der Leber ca. ein Jahr. Freie Fettsäuren werden zur Bildung von VLDL herangezogen; bei Unterernährung oder
Diabetes mellitus (Kohlenhydratmangel der Zellen) zur Bildung von Ketonkörpern (Acetessigsäure,
ß-OH-Buttersäure, Aceton). Diese werden von der Leber nicht benötigt,
sondern dienen der Energieabdeckung extrahepatischer Gewebe (Gehirn
u.a.). Über den Alkoholabbau in der Leber s. dort
Über den Alkoholabbau in der Leber s. dort 
 Die Leber synthetisiert Lipide: Diese benötigt
der Körper als Energielieferanten, Bestandteile der Zellmembran,
Steroide, Gallensäuren, Vitamine. In wässrigen Medien sind Lipide an
Proteine gebunden. Chylomikronen entstehen bei der Fettdigestion und reifen
durch Proteinaustausch zu HDL-Partikeln heran. Leberzellen spalten
in der Resorptionsphase Triglyceride aus HDL, LDL, IDL ab (hepatische Lipase) und verpacken
sie mit ApoB100 zu VLDL-Partikeln, die vor allem in der postresorptiven
Phase für den Energiestoffwechsel genutzt werden. Der Körper
wird in der resorptiven Phase vorwiegend über Chylomikronen, in der
postresorptiven Phase über VLDL mit Triglyceriden versorgt Kurzkettige
Fettsäuren sind gut löslich und werden über die Pfortader
transportiert; langkettige Fettsäuren (>C12), Cholesterin und andere
Lipide in der Darmmukosa in Chylomikronen verpackt. Fettsäuren kann die Leber rasch abbauen, dabei entsteht Acetyl-Coenzym A, das von anderen Geweben verwendet wird; die Leber deckt ihren Energiebedarf hauptsächlich durch Fettsäureoxidation. Aus
resorbierten Kohlenhydraten, Aminosäuren und Fetten bildet die Leber
körpereigene Neutralfette, Phosphoglyzeride, Sphingolipide,
Cholesterin, Gallensäuren, Lezithin. Cholesterin
hemmt seine eigene Synthese über Bremsung der HMG-CoA-Reduktase, regt
Acyltransferase an (Veresterung / Speicherung) und hemmt die
LDL-Rezeptor-Synthese (negative Rückkopplung durch verringerte Aufnahme) In den Zellmembranen einer erwachsenen Person befinden sich
>100 g Cholesterin. Cholesterin aus der Nahrung (~0,3 g/d, steigerbar bis 0,5 g/d) liefert einen Teil
des Bedarfs, den Rest synthetisiert die Leber de novo. Bei Bedarf kann
die Aktivität der HMG-CoA-Reduktase bis ~200-fach ansteigen.
Cholesterin bindet an LDL-Rezeptoren und wird so aus der Blutbahn
entfernt. Die Leber packt Cholesterin in VLDL / HDL-Partikel, die Peripherie kann es in dieser Form verwerten;
überschüssiges Cholesterin gelangt zur Leber zurück (reverser Transport
) Bei Substratmangel dienen Ketonkörper als alternative Energiequelle: Acetessigsäure kann von Gehirn, Muskeln und Nieren (nicht von der Leber
selbst) für deren Energiestoffwechsel verwendet werden (mitochondriale Metabolisierung: Ketonkörper werden zu Acetyl-CoA rückverwandelt und in den Citratzyklus eingeschleust) Hepatozyten
bilden die primären Gallensalze Cholat und Chenodesoxycholat, die mit
Glyzin, Sulfat, Glukuronat oder Taurin konjugiert und in die
Primärgalle der Gallenkanälchen ausgeschieden werden (Lebergalle). Die Gallenblase reduziert durch Rückresorption von Wasser, Kochsalz und Bicarbonat das Volumen der Gallenflüssigkeit isoosmotisch um einen Faktor 10-20, so entsteht Blasengalle (20-50 ml/d, pH 6-7) mit hoher Konzentration an Gallensäuren, Phospholipiden, Cholesterin und Bilirubin. Cholesterin
fällt umso eher aus, je niedriger die Konzentration an Gallensäuren und je höher diejenige an Lezithin ist. CCK führt zur Kontraktion der Gallenblase. Gallensalze können 10-20mal rezirkulieren (Gallensäurepool), sie wirken fettlösend, ohne sie reduziert sich die Fettresorption auf etwa die Hälfte
Die Leber speichert alle fettlöslichen Vitamine (A, D, E, K), diese
werden über Chylomikronen aufgenommen und vor allem in Stellatumzellen
gespeichert Die Leber synthetisiert Lipide: Diese benötigt
der Körper als Energielieferanten, Bestandteile der Zellmembran,
Steroide, Gallensäuren, Vitamine. In wässrigen Medien sind Lipide an
Proteine gebunden. Chylomikronen entstehen bei der Fettdigestion und reifen
durch Proteinaustausch zu HDL-Partikeln heran. Leberzellen spalten
in der Resorptionsphase Triglyceride aus HDL, LDL, IDL ab (hepatische Lipase) und verpacken
sie mit ApoB100 zu VLDL-Partikeln, die vor allem in der postresorptiven
Phase für den Energiestoffwechsel genutzt werden. Der Körper
wird in der resorptiven Phase vorwiegend über Chylomikronen, in der
postresorptiven Phase über VLDL mit Triglyceriden versorgt Kurzkettige
Fettsäuren sind gut löslich und werden über die Pfortader
transportiert; langkettige Fettsäuren (>C12), Cholesterin und andere
Lipide in der Darmmukosa in Chylomikronen verpackt. Fettsäuren kann die Leber rasch abbauen, dabei entsteht Acetyl-Coenzym A, das von anderen Geweben verwendet wird; die Leber deckt ihren Energiebedarf hauptsächlich durch Fettsäureoxidation. Aus
resorbierten Kohlenhydraten, Aminosäuren und Fetten bildet die Leber
körpereigene Neutralfette, Phosphoglyzeride, Sphingolipide,
Cholesterin, Gallensäuren, Lezithin. Cholesterin
hemmt seine eigene Synthese über Bremsung der HMG-CoA-Reduktase, regt
Acyltransferase an (Veresterung / Speicherung) und hemmt die
LDL-Rezeptor-Synthese (negative Rückkopplung durch verringerte Aufnahme) In den Zellmembranen einer erwachsenen Person befinden sich
>100 g Cholesterin. Cholesterin aus der Nahrung (~0,3 g/d, steigerbar bis 0,5 g/d) liefert einen Teil
des Bedarfs, den Rest synthetisiert die Leber de novo. Bei Bedarf kann
die Aktivität der HMG-CoA-Reduktase bis ~200-fach ansteigen.
Cholesterin bindet an LDL-Rezeptoren und wird so aus der Blutbahn
entfernt. Die Leber packt Cholesterin in VLDL / HDL-Partikel, die Peripherie kann es in dieser Form verwerten;
überschüssiges Cholesterin gelangt zur Leber zurück (reverser Transport
) Bei Substratmangel dienen Ketonkörper als alternative Energiequelle: Acetessigsäure kann von Gehirn, Muskeln und Nieren (nicht von der Leber
selbst) für deren Energiestoffwechsel verwendet werden (mitochondriale Metabolisierung: Ketonkörper werden zu Acetyl-CoA rückverwandelt und in den Citratzyklus eingeschleust) Hepatozyten
bilden die primären Gallensalze Cholat und Chenodesoxycholat, die mit
Glyzin, Sulfat, Glukuronat oder Taurin konjugiert und in die
Primärgalle der Gallenkanälchen ausgeschieden werden (Lebergalle). Die Gallenblase reduziert durch Rückresorption von Wasser, Kochsalz und Bicarbonat das Volumen der Gallenflüssigkeit isoosmotisch um einen Faktor 10-20, so entsteht Blasengalle (20-50 ml/d, pH 6-7) mit hoher Konzentration an Gallensäuren, Phospholipiden, Cholesterin und Bilirubin. Cholesterin
fällt umso eher aus, je niedriger die Konzentration an Gallensäuren und je höher diejenige an Lezithin ist. CCK führt zur Kontraktion der Gallenblase. Gallensalze können 10-20mal rezirkulieren (Gallensäurepool), sie wirken fettlösend, ohne sie reduziert sich die Fettresorption auf etwa die Hälfte
Die Leber speichert alle fettlöslichen Vitamine (A, D, E, K), diese
werden über Chylomikronen aufgenommen und vor allem in Stellatumzellen
gespeichert |


