



 Regulation der Calciumhomöostase
Regulation der Calciumhomöostase

| Extrazelluläres Calcium ist für mehrere enzymatische Systeme - wie die Blutgerinnung
- unverzichtbar und stabilisiert das zelluläre Membranpotential. Sinkt
die Konzentration ionalen Calciums in der interstitiellen Flüssigkeit,
dann nimmt die Erregbarkeit von Nerven- und Muskelzellen zu - die
Konzentration an
freiem Ca++ muss über 0,7 mM/l betragen, sonst besteht die Gefahr tetanischer Krämpfe. Die wichtigsten hormonellen Faktoren, welche den Calcium- und Phosphatspiegel steuern, sind -- Parathormon - es wird bei sinkendem Calciumspiegel aus den Epithelkörperchen (Nebenschilddrüsen) freigesetzt, stimuliert Osteoklasten und hebt [Ca++] im Blut -- Calcitonin - parafollikuläre Zellen der Schilddrüse setzen es bei steigendem Calciumspiegel frei; Calcitonin fördert die Mineralisierung des Knochens -- Calcitriol ("Vitamin-D-Hormon") vermehrt die Calciumresorption im Darm. An Osteoblasten regt es die Bildung von Osteocalcin, sowie die Mineralisierung im Knochen an. Im proximalen Nierentubulus hemmt es zusammen mit Parathormon die Ausscheidung von Calcium und Phosphat, der Ca++-und Phosphatspiegel steigt im Blut an -- Der aus dem Knochen stammende Fibroblasten-Wachstumsfaktor 23 (FGF23) führt zu vermehrter Phosphatausscheidung mit dem Harn. Calcitriol wird von Leber und Nieren aus einer (mit der Nahrung aufgenommenen oder in der Haut unter UV-Wirkung entstandenen) Vorstufe in die aktive Form gebracht. Der Aktivierungsschritt in der Niere unterliegt mehrfacher Rückkopplung: Er wird durch Parathormon und abnehmenden Phosphatspiegel im Blut angeregt, durch Calcitriol gehemmt. |
 Parathormon Calcitonin Calcitriol Integrierte Antwort von Parathormon und Calcitriol auf Hypocalcämie
FGF23 Steroidhormone
Parathormon Calcitonin Calcitriol Integrierte Antwort von Parathormon und Calcitriol auf Hypocalcämie
FGF23 Steroidhormone

 Core messages
Core messages
 Abbildung: Bildung und Freisetzung von Parathormon in den Nebenschilddrüsen
Abbildung: Bildung und Freisetzung von Parathormon in den Nebenschilddrüsen Calciumsensitive Rezeptoren (CaSR, calcium-sensing receptors) sind GPC-Rezeptoren, die auf Veränderungen des extrazellulären Calciumspiegels im Bereich von 1-10 mM ansprechen (Normalwert 2,5 mM). In Hauptzellen der Epithelkörperchen hemmen CaSR bei Erhöhung des extrazellulären [Ca++] die Exozytose von Parathormon, in
Tubulusepithelzellen der Niere hemmen sie die Calciumresorption, in Inselzellen des Pankreas wirken sie auf potentialgesteuerte Calciumkanäle und cadheringesteuerte interzelluläre Adhäsion.
Calciumsensitive Rezeptoren (CaSR, calcium-sensing receptors) sind GPC-Rezeptoren, die auf Veränderungen des extrazellulären Calciumspiegels im Bereich von 1-10 mM ansprechen (Normalwert 2,5 mM). In Hauptzellen der Epithelkörperchen hemmen CaSR bei Erhöhung des extrazellulären [Ca++] die Exozytose von Parathormon, in
Tubulusepithelzellen der Niere hemmen sie die Calciumresorption, in Inselzellen des Pankreas wirken sie auf potentialgesteuerte Calciumkanäle und cadheringesteuerte interzelluläre Adhäsion. Knochen (Speicher), Darm und Niere (Resorption und Ausscheidung),
Niere und Leber (Hormonaktivierung und -abbau) sowie die Epithelkörperchen
der Schilddrüse (Hormonsynthese) (Abbildung).
Knochen (Speicher), Darm und Niere (Resorption und Ausscheidung),
Niere und Leber (Hormonaktivierung und -abbau) sowie die Epithelkörperchen
der Schilddrüse (Hormonsynthese) (Abbildung).
 Parathormon aus "Hauptzellen" der Epithelkörperchen (glandulae parathyreoideae
Parathormon aus "Hauptzellen" der Epithelkörperchen (glandulae parathyreoideae  ) - es steigert den Ca++-Spiegel im Blutplasma, wenn dieser abgesunken ist (Thyreo-) Calcitonin aus den parafollikulären Zellen der Schilddrüse - es fördert den Calciumeinbau in den Knochen Calcitriol (Vitamin D3-Hormon: 1,25-Dihydroxy-Calciferol) - es steigert die Resorption von Ca++ im Darm
) - es steigert den Ca++-Spiegel im Blutplasma, wenn dieser abgesunken ist (Thyreo-) Calcitonin aus den parafollikulären Zellen der Schilddrüse - es fördert den Calciumeinbau in den Knochen Calcitriol (Vitamin D3-Hormon: 1,25-Dihydroxy-Calciferol) - es steigert die Resorption von Ca++ im Darm

 Abbildung: Wie die Niere mit Ca++ umgeht
Abbildung: Wie die Niere mit Ca++ umgeht Die Calciumkonzentration im Blutserum beträgt etwa
2,5 mMol/l (10 mg/dl).
Davon sind knapp 60% freie Ionen (das ist der
biologisch wirksame Anteil), die andere Hälfte gebunden (vor allem an
Eiweiß). führen (
Die Calciumkonzentration im Blutserum beträgt etwa
2,5 mMol/l (10 mg/dl).
Davon sind knapp 60% freie Ionen (das ist der
biologisch wirksame Anteil), die andere Hälfte gebunden (vor allem an
Eiweiß). führen ( Mechanismus s. dort). Verwechslung von Infusionsflüssigkeiten: Calcium-komplexierende
Infusionslösungen (Citrat!) können wegen Gefahr von Krampfauslösung
lebensgefährlich sein. Abbildung), sodass die tägliche
Ausscheidung mit dem Harn nur etwa 50 mg beträgt (Vergleich: mit der
Ernährung tägliche Zufuhr etwa 1 Gramm). Wirkung an Rezeptoren Wirkungen im OrganismusAbbildung oben) synthetisieren Parathormon (84
Aminosäure als Präprohormon, aus dem im
endoplasmatischen Retikulum Pro-PTH und im Golgi-Apparat schließlich
Parathormon wird. Dieses wird in sekretorischen Vesikeln gespeichert
und bei Erniedrigung des extrazellulären [Ca++] freigesetzt.
Mechanismus s. dort). Verwechslung von Infusionsflüssigkeiten: Calcium-komplexierende
Infusionslösungen (Citrat!) können wegen Gefahr von Krampfauslösung
lebensgefährlich sein. Abbildung), sodass die tägliche
Ausscheidung mit dem Harn nur etwa 50 mg beträgt (Vergleich: mit der
Ernährung tägliche Zufuhr etwa 1 Gramm). Wirkung an Rezeptoren Wirkungen im OrganismusAbbildung oben) synthetisieren Parathormon (84
Aminosäure als Präprohormon, aus dem im
endoplasmatischen Retikulum Pro-PTH und im Golgi-Apparat schließlich
Parathormon wird. Dieses wird in sekretorischen Vesikeln gespeichert
und bei Erniedrigung des extrazellulären [Ca++] freigesetzt.  Abbildung: Calciumsensor und Regulation des Calciumspiegels
Abbildung: Calciumsensor und Regulation des Calciumspiegels Nach der Transkription von Präproparathormon (115 Aminosäuren) entsteht im endoplasmatischen Retikulum Proparathormon
(90 Aminosäuren). Im Golgi-Apparat werden 6 weitere Aminosäuren
abgespalten, es entsteht Parathormon (84 Aminosäuren). Dieses wird
anschließend in sekretorischen Granula gespeichert, bis es auf Reizung
der Zelle hin freigesetzt wird. Die Expression des Parathormon-Gens wird durch Calcitriol (Vit. D3) unterdrückt:
Nach der Transkription von Präproparathormon (115 Aminosäuren) entsteht im endoplasmatischen Retikulum Proparathormon
(90 Aminosäuren). Im Golgi-Apparat werden 6 weitere Aminosäuren
abgespalten, es entsteht Parathormon (84 Aminosäuren). Dieses wird
anschließend in sekretorischen Granula gespeichert, bis es auf Reizung
der Zelle hin freigesetzt wird. Die Expression des Parathormon-Gens wird durch Calcitriol (Vit. D3) unterdrückt: 
 Hypocalcämie (Werte unter 1,25 mM Ca++) führt innerhalb von Minuten zur Freisetzung von
Parathormon aus den Epithelkörperchen der Nebenschilddrüsen (ggl.
parathyreoideae, Abbildung). Längerdauernder Calciummangel führt zu Hyperplasie der Epithelkörperchen.
Hypocalcämie (Werte unter 1,25 mM Ca++) führt innerhalb von Minuten zur Freisetzung von
Parathormon aus den Epithelkörperchen der Nebenschilddrüsen (ggl.
parathyreoideae, Abbildung). Längerdauernder Calciummangel führt zu Hyperplasie der Epithelkörperchen.| Sinkt der Ca++-Spiegel, wird Parathormon sezerniert |
 Umgekehrt stoppt ein Anstieg
extrazellulärer Ca++-Konzentration (Hypercalcämie) die Parathormonausschüttung.
Umgekehrt stoppt ein Anstieg
extrazellulärer Ca++-Konzentration (Hypercalcämie) die Parathormonausschüttung.  Abbildung: Physiologische Kennkurve der ParathormonregulationAbbildung ganz oben), der Serumspiegel minimal (grüne
Linien). Sinkt der Calciumspiegel, wird die Hemmung der
Parathormonproduktion zusehends aufgehoben, der Hormonspiegel im Blut
steigt. Abbildung).
Abbildung: Physiologische Kennkurve der ParathormonregulationAbbildung ganz oben), der Serumspiegel minimal (grüne
Linien). Sinkt der Calciumspiegel, wird die Hemmung der
Parathormonproduktion zusehends aufgehoben, der Hormonspiegel im Blut
steigt. Abbildung).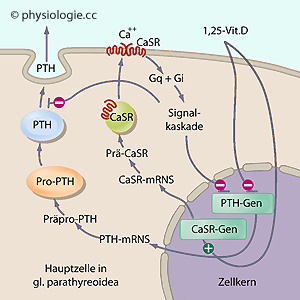 Abbildung: Regulation der Expression von CaSR und PTH, und der Sekretion von PTH In den Epithelkörperchen der Schilddrüse: CaSR (Abbildung rechts und oben) bindet an
der Membran-Außenseite Ca++-Ionen. Dies wirkt sich über Gq- und Gi-Proteine aus: Gq-Proteine aktivieren über den PLC-IP3-Ca++-Weg Proteinkinase C, dies führt zu Freisetzung von Ca++ aus
intrazellulären Speichern. Der steigende Calciumspiegel in der Zelle
hemmt die Sekretion bereits gebildeten Parathormons aus
Speichergranula der Epithelkörperchenzellen und inhibiert die Expression des PTH-Gens im Zellkern. Es wird also weniger Parathormon gebildet und das bereits gespeicherte wird nicht exozytiert. Hier führt ausnahmsweise eine Steigerung des intrazellulären [Ca++] nicht zur Anregung, sondern zu einer Hemmung der Vesikelfusion und Sekretion (ähnlich wie in der Niere, wo intrazellulärer Ca++-Anstieg die Freisetzung von Renin hemmt). Die Aktivierung von Gi hemmt Adenylylcyclase,
[cAMP] nimmt ab, die Aktivität der Proteinkinase A sinkt, die
Transkription des Parathormon-Gens sowie die Exozytose von Vesikeln ebenfalls.
Abbildung: Regulation der Expression von CaSR und PTH, und der Sekretion von PTH In den Epithelkörperchen der Schilddrüse: CaSR (Abbildung rechts und oben) bindet an
der Membran-Außenseite Ca++-Ionen. Dies wirkt sich über Gq- und Gi-Proteine aus: Gq-Proteine aktivieren über den PLC-IP3-Ca++-Weg Proteinkinase C, dies führt zu Freisetzung von Ca++ aus
intrazellulären Speichern. Der steigende Calciumspiegel in der Zelle
hemmt die Sekretion bereits gebildeten Parathormons aus
Speichergranula der Epithelkörperchenzellen und inhibiert die Expression des PTH-Gens im Zellkern. Es wird also weniger Parathormon gebildet und das bereits gespeicherte wird nicht exozytiert. Hier führt ausnahmsweise eine Steigerung des intrazellulären [Ca++] nicht zur Anregung, sondern zu einer Hemmung der Vesikelfusion und Sekretion (ähnlich wie in der Niere, wo intrazellulärer Ca++-Anstieg die Freisetzung von Renin hemmt). Die Aktivierung von Gi hemmt Adenylylcyclase,
[cAMP] nimmt ab, die Aktivität der Proteinkinase A sinkt, die
Transkription des Parathormon-Gens sowie die Exozytose von Vesikeln ebenfalls. Sinkender Ca++-Spiegel im Blut stimuliert, steigender hemmt die Freisetzung von Parathormon.
Auch Tubuluszellen in der Niere (sie regulieren die Rückresorption / renale Ausscheidung von Calcium und Magnesium) und Zellen im Knochen verfügen über
CaSR.
Sinkender Ca++-Spiegel im Blut stimuliert, steigender hemmt die Freisetzung von Parathormon.
Auch Tubuluszellen in der Niere (sie regulieren die Rückresorption / renale Ausscheidung von Calcium und Magnesium) und Zellen im Knochen verfügen über
CaSR. In den Kreislauf freigesetztes Parathormon wird rasch abgebaut - hauptsächlich (90%) von Leber und Nieren (Halbwertszeit 2-5 Minuten). Durch die Abtrennung eines N-terminalen Fragments verliert das Hormon seine biologische Wirksamkeit.
In den Kreislauf freigesetztes Parathormon wird rasch abgebaut - hauptsächlich (90%) von Leber und Nieren (Halbwertszeit 2-5 Minuten). Durch die Abtrennung eines N-terminalen Fragments verliert das Hormon seine biologische Wirksamkeit. Am häufigsten findet sich der "klassische" PTH1R
an Zellen in den Nieren (Tubulusepithel) und Knochen (Osteoblasten).
Aktivierung von PTH1R auf Osteoblasten führt zu deren Expression von RANKL, dieses koppelt auf RANK auf Osteoklasten-Vorläuferzellen und verwandelt sie zu aktiven Osteoklasten (Knochenabbau).Der PTH2R findet sich vor allem in Zentralnervensystem, im Pankreas sowie in Plazenta und Hoden.
Am häufigsten findet sich der "klassische" PTH1R
an Zellen in den Nieren (Tubulusepithel) und Knochen (Osteoblasten).
Aktivierung von PTH1R auf Osteoblasten führt zu deren Expression von RANKL, dieses koppelt auf RANK auf Osteoklasten-Vorläuferzellen und verwandelt sie zu aktiven Osteoklasten (Knochenabbau).Der PTH2R findet sich vor allem in Zentralnervensystem, im Pankreas sowie in Plazenta und Hoden. Abbildung: Parathormonrezeptor Zunächst entsteht ein lockerer
Hormon-Rezeptor-Komplex, der sich durch
Konformationsänderung des Rezeptors festigt und dann die Adenylylcyclase einschaltet. Nach Internalisierung des Komplexes folgt
eine länger anhaltende Phase, die cAMP-Konzentration in der Zelle
bleibt erhöht ( Abbildung).Liganden mit höherer Affinität zur RG-Konformation
des Rezeptors bewirken eher rasche, anabole Effekte am Knochen.
(Wiederholte Parathormongaben niedriger Dosierung fördern den
Knochenaufbau.)Liganden mit höherer Affinität zu R0
wirken über längere Dauer auf den Knochen resorptionsfördernd.
(Konstant erhöhte Parathormonspiegel fördern die Resorption von
Knochensubstanz.) Längerer Anstieg des Parathormonspiegels fördert den Abbau von Knochensubstanz - die Resorption indirekt,
denn Osteoklasten haben keine PTH-Rezeptoren, Osteoblasten und
Osteoklasten-Vorläufer schon. Letztere produzieren auf das PTH-Signal
hin Zytokine, die Osteoklasten anregen. Kurzzeitiger Anstieg des Parathormonspiegels regt vor allem die Neubildung von Knochensubstanz an. Dies erfolgt direkt durch Öffnung von Calciumkanälen in Osteozyten, was Ca++ aus der extrazellulären Knochenflüssigkeit in die Zellen lässt - von dort gelangt es über gap junctions in Osteoblasten an der Oberfläche der Knochenstruktur (osteozytische Osteolyse).
Die Osteoblasten deponieren dieses Calcium anschließend in die
Knochenmatrix. Auch senkt Parathormon die Bildung von Sclerostin in
Osteozyten, was die Osteoblasten schützt. Parathormon steigert die renale Calciumresorption im dicken aufsteigenden Schenkel der Henle-Schleife (TAL) sowie in der pars convoluta des distalen Tubulus ([Ca++] steigt im Blut, sinkt in Harn) - vermutlich zur Gänze indirekt, indem es die Synthese von 1,25-(OH2)-Vit.D fördert.
Parathormon hemmt die Phosphatresorption im proximalen Tubulus, insbesondere bei ausreichendem Phosphatangebot mit der Ernährung ([Phosphat] steigt im Harn - Phosphaturie -, sinkt im Blut). Die vermehrte Ausscheidung von
Phosphat erfolgt durch Verlagerung von Na/Phosphat-Cotransportern aus
der apikalen Membran von Tubulusepithelzellen in den Pool
intrazellulärer Vesikel.
Abbildung: Parathormonrezeptor Zunächst entsteht ein lockerer
Hormon-Rezeptor-Komplex, der sich durch
Konformationsänderung des Rezeptors festigt und dann die Adenylylcyclase einschaltet. Nach Internalisierung des Komplexes folgt
eine länger anhaltende Phase, die cAMP-Konzentration in der Zelle
bleibt erhöht ( Abbildung).Liganden mit höherer Affinität zur RG-Konformation
des Rezeptors bewirken eher rasche, anabole Effekte am Knochen.
(Wiederholte Parathormongaben niedriger Dosierung fördern den
Knochenaufbau.)Liganden mit höherer Affinität zu R0
wirken über längere Dauer auf den Knochen resorptionsfördernd.
(Konstant erhöhte Parathormonspiegel fördern die Resorption von
Knochensubstanz.) Längerer Anstieg des Parathormonspiegels fördert den Abbau von Knochensubstanz - die Resorption indirekt,
denn Osteoklasten haben keine PTH-Rezeptoren, Osteoblasten und
Osteoklasten-Vorläufer schon. Letztere produzieren auf das PTH-Signal
hin Zytokine, die Osteoklasten anregen. Kurzzeitiger Anstieg des Parathormonspiegels regt vor allem die Neubildung von Knochensubstanz an. Dies erfolgt direkt durch Öffnung von Calciumkanälen in Osteozyten, was Ca++ aus der extrazellulären Knochenflüssigkeit in die Zellen lässt - von dort gelangt es über gap junctions in Osteoblasten an der Oberfläche der Knochenstruktur (osteozytische Osteolyse).
Die Osteoblasten deponieren dieses Calcium anschließend in die
Knochenmatrix. Auch senkt Parathormon die Bildung von Sclerostin in
Osteozyten, was die Osteoblasten schützt. Parathormon steigert die renale Calciumresorption im dicken aufsteigenden Schenkel der Henle-Schleife (TAL) sowie in der pars convoluta des distalen Tubulus ([Ca++] steigt im Blut, sinkt in Harn) - vermutlich zur Gänze indirekt, indem es die Synthese von 1,25-(OH2)-Vit.D fördert.
Parathormon hemmt die Phosphatresorption im proximalen Tubulus, insbesondere bei ausreichendem Phosphatangebot mit der Ernährung ([Phosphat] steigt im Harn - Phosphaturie -, sinkt im Blut). Die vermehrte Ausscheidung von
Phosphat erfolgt durch Verlagerung von Na/Phosphat-Cotransportern aus
der apikalen Membran von Tubulusepithelzellen in den Pool
intrazellulärer Vesikel.| Bei Parathormonmangel (Hypoparathyreoidismus) steigt der Plasma-Phosphatspiegel |
Parathormon steigert die Synthese von aktivem Vitamin-D-Hormon (Calcitriol) in der Niere. Es regt in renalen Mitochondrien über Aktivierung renalen 1α-Hydroxylase CYP 27B1 an ( s. weiter unten). Calcitriolbildend wirken weiters ein Absinken des Ca++- und Phosphatspiegels im Serum sowie Östrogene und Prolaktin.| Parathormon regt die renale Calcitriolbildung an |

 Abbildung: Regulierung des Calciums durch Parathormon und Calcitriol (=Vitamin D3)
Abbildung: Regulierung des Calciums durch Parathormon und Calcitriol (=Vitamin D3)| PTH mobilisiert Calciumphosphat aus dem Knochen. In der Niere fördert es Calciumresorption und Phosphatausscheidung |
Rückkopplungskreis: Erhöhtes [Ca++] hemmt die Freisetzung von Parathormon; der fallende Parathormonspiegel senkt die Vit-D-Synthese in der Niere; die Ca++-Resorption nimmt ab; der Ca++-Spiegel fällt; das wiederum stimuliert die Parathormonproduktion; Parathormon steigert den Ca++-Spiegel; etc. Abbildung: Rückkopplungsschleifen der Calcium- und Phosphat-Homöostase Die gesteigerte Phosphatausscheidung verhindert, dass es außerhalb des Knochens zum Ausfällen von Calciumphosphat kommt.
Ein erniedrigter Phosphatspiegel beugt einem Überschreiten des Löslichkeitsprodukts im Gewebe - und einem Ausfall von Calciumphosphatkristallen - vor.). Dies kann die Folge einer Überproduktion von Parathormon (primärer Hyperparathyreoidismus) sein. Wenn größere Steine in den Ureter gelangen und
diesen verlegen, kann die reaktive Kontraktion der glatten Muskulatur
zu Durchblutungsabnahme und Sauerstoffmangel an der betreffenden Stelle
führen. Eine daraus resultierende Nierenkolik verursacht überaus heftige Schmerzen.
Abbildung: Rückkopplungsschleifen der Calcium- und Phosphat-Homöostase Die gesteigerte Phosphatausscheidung verhindert, dass es außerhalb des Knochens zum Ausfällen von Calciumphosphat kommt.
Ein erniedrigter Phosphatspiegel beugt einem Überschreiten des Löslichkeitsprodukts im Gewebe - und einem Ausfall von Calciumphosphatkristallen - vor.). Dies kann die Folge einer Überproduktion von Parathormon (primärer Hyperparathyreoidismus) sein. Wenn größere Steine in den Ureter gelangen und
diesen verlegen, kann die reaktive Kontraktion der glatten Muskulatur
zu Durchblutungsabnahme und Sauerstoffmangel an der betreffenden Stelle
führen. Eine daraus resultierende Nierenkolik verursacht überaus heftige Schmerzen. | Primärer Hyperparathyreoidismus kann zu Nephrolithiasis (Bildung von Nierensteinen) führen |
Die Gesamtwirkung des Parathormons ist eine (wenn auch kurzfristige) Steigerung der Ca++-Konzentration im Blut; im Harn
nimmt die Konzentration von Calcium ab, die
von Phosphat zu. (Die Menge an ausgeschiedenem Calcium steigt über längere Zeit im Endeffekt an.) Auch fördert Parathormon die Resorption von Magnesium in der Niere (Henle-Schleife, distaler Tubulus). Die Mobilisierung von Calcium aus dem Knochen würde über längere Zeit zu dessen
Entkalkung führen. Parathormon regt aber auch die Aktivierung von Calcitriol in der Niere an (Induktion der 1α-Hydroxylase). Dies fördert die Calcium- und Phosphataufnahme im Darm. Das ist wichtig, weil die längerfristige Wirkung des Parathormons - im Gegensatz zum Kurzzeiteffekt - eine vermehrte Ausscheidung von Calciumphosphat
ist, wegen der erhöhten Menge an mobilisiertem Salz, das unter
PTH-Wirkung aus dem Knochen gerät und schließlich mit dem Harn entfernt
wird.Abbildung).
Die Mobilisierung von Calcium aus dem Knochen würde über längere Zeit zu dessen
Entkalkung führen. Parathormon regt aber auch die Aktivierung von Calcitriol in der Niere an (Induktion der 1α-Hydroxylase). Dies fördert die Calcium- und Phosphataufnahme im Darm. Das ist wichtig, weil die längerfristige Wirkung des Parathormons - im Gegensatz zum Kurzzeiteffekt - eine vermehrte Ausscheidung von Calciumphosphat
ist, wegen der erhöhten Menge an mobilisiertem Salz, das unter
PTH-Wirkung aus dem Knochen gerät und schließlich mit dem Harn entfernt
wird.Abbildung).  Abbildung: Parafollikuläre Zellen Calcitonin fördert die Mineralisierung des Knochens, indem es die Osteolyse bremst - über rezeptorvermittelte
Inhibition der Osteoklasten (die reich an Calcitoninrezeptoren sind)
und Anregung der Osteoblasten. Diese Wirkungen klingen rasch ab (innerhalb weniger Stunden dauerhaft erhöhten Calcitoninspiegels), sie sind transitorisch.
Abbildung: Parafollikuläre Zellen Calcitonin fördert die Mineralisierung des Knochens, indem es die Osteolyse bremst - über rezeptorvermittelte
Inhibition der Osteoklasten (die reich an Calcitoninrezeptoren sind)
und Anregung der Osteoblasten. Diese Wirkungen klingen rasch ab (innerhalb weniger Stunden dauerhaft erhöhten Calcitoninspiegels), sie sind transitorisch.| Calcitonin reduziert die Osteoklasten-Aktivität |
Calcitonin verringert bei niedrigem Serum-[Ca++] die
Rückresorption von Phosphat im proximalen Nierentubulus und steigert die Rückresorption von Calcium im distalen Tubulus. In dieser Hinsicht wirkt Calcitonin synergistisch mit Parathormon, das ebenfalls die renale Rückresorption von Ca++ und Sekretion von HPO4-- fördert. Calcitonin regt die Sclerostinbildung
der Osteozyten an, was antianabol auf den Knochen wirkt. Sclerostin ist
ein Glykoprotein, das die knochenbildende Wirkung der Osteoblasten
hemmt. Calcitonin hemmt
bei Hypercalcämie kurzfristig die Bildung von Parathormon, wirkt aber
über längere Zeiträume synergistisch im Sinne von Knochenaufbau und
-erhalt (vor allem bei Wachstum und Laktation). Calcitonin hat außerdem (wahrscheinlich durch Endorphinfreisetzung) eine analgetische Wirkung im ZNS. Calcitonin s. dort
Calcitonin s. dort 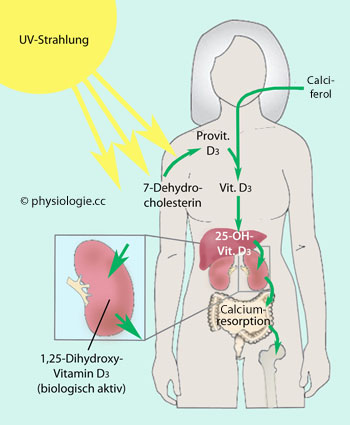 Abbildung: Endogene Vitamin-D-Synthese
Abbildung: Endogene Vitamin-D-Synthese Abbildung: Vitamin D-Rezeptor Calcitriol induziert Calbindin, ein u.a. vom Darmepithel exprimiertes Ca++-Bindungs-
und Transportprotein. Dieser Mechanismus verlängert die Wirkungszeit
des Vitamin-D-Hormons und verbessert die Aufnahme des Calciums aus der
Nahrung (erhöhter Nutzungsgrad). Calcitriol erhöht die Ca++- (synergistisch mit Parathormon) und Phosphat-Resorption in der Niere (Henle-Schleife, distaler Tubulus). Calcitriol hemmt weiters die renale 1-Hydroxylierung des Vit.D und damit seine eigene Synthese (negatives Feedback). Calcitriol fördert die Calcium- und die Phosphatresorption im Dünndarm - letzteres
über Na/P-Cotransporter, welche den geschwindigkeitslimitierenden
Schritt für den transepithelialen Transport darstellen. Calcitriol wirkt größtenteils indirekt auf den Knochen - über seinen Einfluss auf Calcium- und Phosphattransport in Darm und Nieren. Direkt bewirkt es an den Osteoblasten bei ausreichendem [Ca++] die Synthese von Matrixmaterial und die Mineralisierung; bei Hypocalcämie fördert es über Osteoklasten den Calciumabbau. durch Parathormon und abnehmenden Calcium- bzw. Phosphatspiegel im Blut angeregt und durch Calctriol (Selbsthemmung) sowie Hypercalcämie und Hyperphosphatämie inhibiert.
Abbildung: Vitamin D-Rezeptor Calcitriol induziert Calbindin, ein u.a. vom Darmepithel exprimiertes Ca++-Bindungs-
und Transportprotein. Dieser Mechanismus verlängert die Wirkungszeit
des Vitamin-D-Hormons und verbessert die Aufnahme des Calciums aus der
Nahrung (erhöhter Nutzungsgrad). Calcitriol erhöht die Ca++- (synergistisch mit Parathormon) und Phosphat-Resorption in der Niere (Henle-Schleife, distaler Tubulus). Calcitriol hemmt weiters die renale 1-Hydroxylierung des Vit.D und damit seine eigene Synthese (negatives Feedback). Calcitriol fördert die Calcium- und die Phosphatresorption im Dünndarm - letzteres
über Na/P-Cotransporter, welche den geschwindigkeitslimitierenden
Schritt für den transepithelialen Transport darstellen. Calcitriol wirkt größtenteils indirekt auf den Knochen - über seinen Einfluss auf Calcium- und Phosphattransport in Darm und Nieren. Direkt bewirkt es an den Osteoblasten bei ausreichendem [Ca++] die Synthese von Matrixmaterial und die Mineralisierung; bei Hypocalcämie fördert es über Osteoklasten den Calciumabbau. durch Parathormon und abnehmenden Calcium- bzw. Phosphatspiegel im Blut angeregt und durch Calctriol (Selbsthemmung) sowie Hypercalcämie und Hyperphosphatämie inhibiert.
 Aufnahme von Calcium und Phosphat im Darm ( s. dort): Calcitriol beeinflusst die transzelluläre Resorption von Ca++ durch Mukosazellen des Duodenums. Das betrifft drei Schritte: Passage von Calciumionen durch die apikale Membran via TRPV6-Kanäle Intrazelluläre Kopplung an Bindungsproteine, insbesondere Calbindin (puffert Ca++-Konzentrationsschwankungen und erhält so den Konzentrationsgradienten für Calciumeinstrom über die apikale Membran) Passage von Calciumionen über die basolaterale Membran via Calciumpumpen und Na/Ca-Austauscher Osteoblasten: Reifung und Anregung der Bildung von Osteocalcin sowie Bildung von Hydroxylapatitkristallen (Mineralisierung) im Knochen, wo Calcitriol gleichzeitig die Reifung und Resorptionstätigkeit der Osteoklasten stimuliert und dadurch den Umbau des Knochens beschleunigt Im proximalen Tubulus der Niere wird die Ca++- und HPO4---Ausscheidung gehemmt (allerdings nur, wenn gleichzeitig Parathormon wirkt) - das erhöht den Ca++- und HPO4---spiegel im Blut Calcitriol unterdrückt die Expression des Parathormon-Gens Zu Wirkungen des Vitamin D, die nichts mit dem Calciumstoffwechsel zu tun haben,
gehören Reifung und Differenzierung zahlreicher Zellen, sowie die Zytokinbildung - Vit.D ist also auch für die Immunabwehr bedeutsam.
Aufnahme von Calcium und Phosphat im Darm ( s. dort): Calcitriol beeinflusst die transzelluläre Resorption von Ca++ durch Mukosazellen des Duodenums. Das betrifft drei Schritte: Passage von Calciumionen durch die apikale Membran via TRPV6-Kanäle Intrazelluläre Kopplung an Bindungsproteine, insbesondere Calbindin (puffert Ca++-Konzentrationsschwankungen und erhält so den Konzentrationsgradienten für Calciumeinstrom über die apikale Membran) Passage von Calciumionen über die basolaterale Membran via Calciumpumpen und Na/Ca-Austauscher Osteoblasten: Reifung und Anregung der Bildung von Osteocalcin sowie Bildung von Hydroxylapatitkristallen (Mineralisierung) im Knochen, wo Calcitriol gleichzeitig die Reifung und Resorptionstätigkeit der Osteoklasten stimuliert und dadurch den Umbau des Knochens beschleunigt Im proximalen Tubulus der Niere wird die Ca++- und HPO4---Ausscheidung gehemmt (allerdings nur, wenn gleichzeitig Parathormon wirkt) - das erhöht den Ca++- und HPO4---spiegel im Blut Calcitriol unterdrückt die Expression des Parathormon-Gens Zu Wirkungen des Vitamin D, die nichts mit dem Calciumstoffwechsel zu tun haben,
gehören Reifung und Differenzierung zahlreicher Zellen, sowie die Zytokinbildung - Vit.D ist also auch für die Immunabwehr bedeutsam. Abbildung: Ultraviolett A, B und CAbbildung). Abbildung).
Abbildung: Ultraviolett A, B und CAbbildung). Abbildung). Abbildung: Vitamin-D-Synthese, solche von CYP 24A1 zu D-Hypervitaminose. In der Haut wird unter UV-Wirkung 7-Dehydrocholesterin zu Prävitamin D3 umgewandelt und dieses isomerisiert zu Cholecalciferol. Die Leber bildet daraus (mittels mitochondrialer und mikrosomaler Enzyme) Calcidiol; dieses wird in der Niere
(mittels 1α-Hydroxylase) zu Calcitriol (Vit-D-Hormon) umgewandelt
( Abbildung). Dieser Schritt erfolgt in den Mitochondrien des
proximalen Tubulus und wird durch Parathormon, Hypophosphatämie, aber auch durch Prolaktin angeregt.
Abbildung: Vitamin-D-Synthese, solche von CYP 24A1 zu D-Hypervitaminose. In der Haut wird unter UV-Wirkung 7-Dehydrocholesterin zu Prävitamin D3 umgewandelt und dieses isomerisiert zu Cholecalciferol. Die Leber bildet daraus (mittels mitochondrialer und mikrosomaler Enzyme) Calcidiol; dieses wird in der Niere
(mittels 1α-Hydroxylase) zu Calcitriol (Vit-D-Hormon) umgewandelt
( Abbildung). Dieser Schritt erfolgt in den Mitochondrien des
proximalen Tubulus und wird durch Parathormon, Hypophosphatämie, aber auch durch Prolaktin angeregt. | Die Leber bildet aus Cholecalciferol Calcidiol Die Nieren bilden aus Calcidiol mittels 1α-Hydroxylase Calcitriol. Dieser Schritt wird u.a. durch Parathormon angeregt Schwere Niereninsuffizienz bedingt Calcitriolmangel |
Calcitriol
wird hauptsächlich über die Galle ausgeschieden. (Gallenflüssigkeit ist
auch für die Aufnahme des - fettlöslichen - Vitamins erforderlich).| Vitamin-D-Mangel führt kompensatorisch zu erhöhter Parathormonsekretion |
| D-Hypervitaminose lässt das freie Calcium im Serum ansteigen |
Geschlechtshormone unterstützen den Knochenaufbau, Glucocotricoide hingegen den Knochenabbau. Östrogene
regen
den Hydroxylierungsschritt in der Leber an; Östrogenmangel
(postklimakterisch) könnte auch auf diese Weise zur Entwicklung einer
Osteoporose beitragen. Östrogene wirken anabol auf den Knochen. Testosteron ist beim Mann für die Aufrechterhaltung einer normalen Knochendichte ebenfalls unverzichtbar. Gesteigerter Blutspiegel an Glucocorticoiden befördert Osteoporose, was durch Wirkungen auf die Bildung von Osteoprotegerin und RANKL erklärbar ist. Ca und Pi in der Nahrung wirken sich auf mineralwirksame Hormone aus. Der Parathormonspiegel und der Vit-D-Hormon-Spiegel im Blut sinken durch intestinale Calciumaufnahme rwegen des Anstiegs im Serum-[Ca++],
der die Parathormonsekretion hemmt, was wiederum die Calcium- und
Phosphatresorption im Knochen unterdrückt und so den postprandialen
Calcium- und Phosphatanstieg begrenzt. Das Absinken des
Parathormonspiegels reduziert außerdem die Calciumresorption in der
Niere und bewirkt Calciurie. Bei anhaltender Calciumresorption im Darm
senkt der niedrige Parathormonspiegel außerdem die 1-Hydroxylierung des
25-OH-Vit.D, und das senkt wiederum die Calciumresorption im Darm. Der Parathormonspiegel und der Vit-D-Hormon-Spiegel im Blut steigen durch intestinale Aufnahme relativ großer Mengen Phosphat.
In dieser Situation steigt der Phosphatspiegel im Plasma, was im
Knochen zu vermehrter Mineralisierung führt. Calcium wird dabei
verbraucht, [Ca++] im Blutplasma sinkt, die
Parathormonsekretion steigt an, Phosphat wird vermehrt mit dem Harn
ausgeschieden; gleichzeitig wird mehr Calciumphosphat aus dem Knochen
mobilisiert. Der Phosphateffekt funktioniert also indirekt über die
Steigerung des Parathormonspiegels. Bei längerer Wirkungsdauer nimmt
zudem die 1-Hydroxylierung des 25-OH-Vit.D zu, der Vit-D-Hormonspiegel im Blut steigt an (und damit seine Wirkungen).
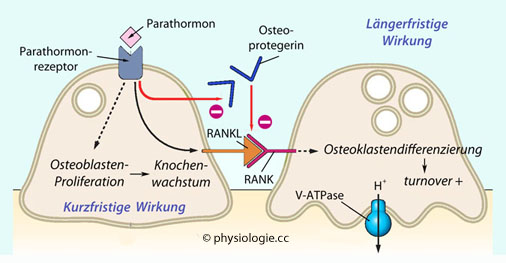 Abbildung: Kurz- vs. Langzeitwirkung des Parathormons auf den Knochen Kurzfristig (Minuten bis
Stunden - Abbildung, links): Absinken des extrazellulären [Ca++] wird von CaSR auf Zellen
der Nebenschilddrüsen registriert und stimuliert die Sekretion von
Parathormon. Parathormon mobilisiert Ca++
aus Niere (vermehrte Rückresorption in distalen Tubuli) und Knochen
(Expression von RANKL auf Osteoblasten, erhöhte Osteoklastenaktivität,
Knochensubstanz wird abgebaut). Der Calcium- und Phosphatspiegel im
Blut steigt an. Auch hemmt Parathormon die renale Phosphatresorption: Es hemmt den natriumabhängigen Phosphattransporter im proximalen Tubulus → vermehrte
Phosphatausscheidung, verringerter Serumspiegel, weniger Bindung freien Calciums. Längerfristig (Stunden bis Tage - Abbildung, rechts): Anhaltende Hypocalcämie aktiviert über Stimulierung der renalen 1α-Hydroxylase Vitamin D3. Das regt die Ca++-Resorption
in Darm und Knochen an (auch die von Phosphat). Die Expression von
TRPV-Calciumkanälen, Calbindin und PMCA - und damit die Kapazität der
Epithelzellen für den Ca++-Import
- nimmt zu. Auch stimuliert Calcitriol die Bildung von RANKL auf
Osteoblasten, was den Parathormon-Effekt auf den Knochen verstärkt. Negative Rückkopplung:
Erhöhte Parathormonspiegel steigern die Synthese von Calcitriol, und
dieses reprimiert direkt die Expression des Parathormon-Gens (gl.
parathyreoidea) sowie die Aktivität der 1α-Hydroxylase (Niere). FGF23 reduziert vor allem die Resorption von Phosphat im proximalen Nierentubulus (es hemmt die Expression von Natrium / Phosphat-Cotransportern und damit deren Einbau in die apikale Membran) und senkt so den Phosphatspiegel. FGF23 erhöht die Calciumresorption aus distalen Nierentubuli, indem es die Expression von TRPV5-Glykoproteinen
(für die Calciumaufnehme wesentliche Membrankanäle) in die distalen
Tubuli anregt. Gleichzeitig senkt es die Sekretion von Parathormon, das
seinerseits die Bildung von FGF anregt. FGF23 reduziert die Bildung von Calcitriol
im proximalen Tubulus durch Hemmung der Synthese (1α-Hydroxylase) und
Förderung des Abbaus (24-Hydroxylase). Umgekehrt fördert Calcitriol die
Bildung von FGF23.
Abbildung: Kurz- vs. Langzeitwirkung des Parathormons auf den Knochen Kurzfristig (Minuten bis
Stunden - Abbildung, links): Absinken des extrazellulären [Ca++] wird von CaSR auf Zellen
der Nebenschilddrüsen registriert und stimuliert die Sekretion von
Parathormon. Parathormon mobilisiert Ca++
aus Niere (vermehrte Rückresorption in distalen Tubuli) und Knochen
(Expression von RANKL auf Osteoblasten, erhöhte Osteoklastenaktivität,
Knochensubstanz wird abgebaut). Der Calcium- und Phosphatspiegel im
Blut steigt an. Auch hemmt Parathormon die renale Phosphatresorption: Es hemmt den natriumabhängigen Phosphattransporter im proximalen Tubulus → vermehrte
Phosphatausscheidung, verringerter Serumspiegel, weniger Bindung freien Calciums. Längerfristig (Stunden bis Tage - Abbildung, rechts): Anhaltende Hypocalcämie aktiviert über Stimulierung der renalen 1α-Hydroxylase Vitamin D3. Das regt die Ca++-Resorption
in Darm und Knochen an (auch die von Phosphat). Die Expression von
TRPV-Calciumkanälen, Calbindin und PMCA - und damit die Kapazität der
Epithelzellen für den Ca++-Import
- nimmt zu. Auch stimuliert Calcitriol die Bildung von RANKL auf
Osteoblasten, was den Parathormon-Effekt auf den Knochen verstärkt. Negative Rückkopplung:
Erhöhte Parathormonspiegel steigern die Synthese von Calcitriol, und
dieses reprimiert direkt die Expression des Parathormon-Gens (gl.
parathyreoidea) sowie die Aktivität der 1α-Hydroxylase (Niere). FGF23 reduziert vor allem die Resorption von Phosphat im proximalen Nierentubulus (es hemmt die Expression von Natrium / Phosphat-Cotransportern und damit deren Einbau in die apikale Membran) und senkt so den Phosphatspiegel. FGF23 erhöht die Calciumresorption aus distalen Nierentubuli, indem es die Expression von TRPV5-Glykoproteinen
(für die Calciumaufnehme wesentliche Membrankanäle) in die distalen
Tubuli anregt. Gleichzeitig senkt es die Sekretion von Parathormon, das
seinerseits die Bildung von FGF anregt. FGF23 reduziert die Bildung von Calcitriol
im proximalen Tubulus durch Hemmung der Synthese (1α-Hydroxylase) und
Förderung des Abbaus (24-Hydroxylase). Umgekehrt fördert Calcitriol die
Bildung von FGF23.| Parathormon, Vit-D-Hormon, FGF23: Wirkungen auf die Ca++ / Pi- Homöostase Nach White / Harrison / Mehlmann, Endocrine and reproductive physiology, 5th ed., Elsevier 2019 |
||||
| |
Dünndarm |
Knochen |
Nieren |
Epithelkörperchen |
| Parathormon |
keine direkte Wirkung |
reguliert Differenzierung / Funktion von Osteoklasten über RANKL / OPG- Expression in Osteoblasten |
stimuliert 1α- Hydroxylase und Produktion von 1,25(OH)2D3 | keine direkte Wirkung |
| 1,25(OH)2D3 |
erhöht Ca++-Resorption über TRPV-Kanäle, Calbindin und Plasmamembran - Ca++ ATPase |
reguliert Differenzierung / Funktion von Osteoklasten über RANKL / OPG- Expression in Osteoblasten | unterstützt Ca++- Resorption im distalen Tubulus über Calbindin unterstützt Phosphat- resorption im proximalen Tubulus |
hemmt Transkription des Parathormon- gens fördert die Expression des CaSR-Gens |
| FGF23 |
keine direkte Wirkung | Osteozyten bilden FGF23 |
hemmt Phosphat- resorption im proximalen Tubulus / 1α-Hydroxylase / Produktion von 1,25(OH)2D3 |
? |
Östradiol regt die Ca++-Resorption
sowohl im Darm als auch in den Nierentubuli an, hebt die Lebensdauer
von Osteoblasten und stimuliert die Apoptose von Osteoklasten. All das
wirkt fördernd auf Knochenwachstum und -dichte. Der postmenopausale
Östrogenabfall bedingt für ~5 Jahre raschen Knochenabbau, dessen Tempo
sich anschließend verlangsamt. Testosteron hat ähnliche Effekte, allerdings zum Teil durch Östradiol, das durch periphere Umwandlung aus Testosteron entsteht. Hochdosierte Glucocorticoide
(Corticoidtherapie, Mb. Cushing) bewirken umgekehrt Knochenabbau und
Osteoporose - über gehemmte Differenzierung und Funktion der
Osteoblasten, vermehrte Resorption von Knochen, verminderte Calciumresorption im Darm sowie erhöhte Calciumausscheidung mit dem
Harn (physiologische Glucocorticoidkonzentrationen haben keine solchen
Effekte). Hormonmangel:
Bei fehlerhafter Anlage (Chromosom-22-Mikrodeletionssyndrom),
Zerstörung oder Entfernung der Epithelkörperchen (Operation,
Bestrahlung) fällt die Parathormonbildung aus. Hormonumenpfindlichkeit: Bei
ungestörter Hormonproduktion, aber mangelnder Empfindlichkeit der
Zielzellen gegenüber Parathormon (Rezeptordefekt, gestörte
second-messenger-Mechanismen) besteht ein Pseudohypoparathyreoidismus.
Hormonmangel:
Bei fehlerhafter Anlage (Chromosom-22-Mikrodeletionssyndrom),
Zerstörung oder Entfernung der Epithelkörperchen (Operation,
Bestrahlung) fällt die Parathormonbildung aus. Hormonumenpfindlichkeit: Bei
ungestörter Hormonproduktion, aber mangelnder Empfindlichkeit der
Zielzellen gegenüber Parathormon (Rezeptordefekt, gestörte
second-messenger-Mechanismen) besteht ein Pseudohypoparathyreoidismus. Abbildung: Pathophysiologie des sekundären Hyperparathyreoidismus Abbildung). Absinken der Calciumkonzentration (Hypocalcämie) erhöht die Erregbarkeit von Zellmembranen, weil
der Natriumeinstrom erleichtert wird, und bewirkt tetanische Krämpfe,
die lebensbedrohlich sein können. Gegenmaßnahme: Erhöhung des Calciumspiegels (Calciumcarbonat i.v.).
Abbildung: Pathophysiologie des sekundären Hyperparathyreoidismus Abbildung). Absinken der Calciumkonzentration (Hypocalcämie) erhöht die Erregbarkeit von Zellmembranen, weil
der Natriumeinstrom erleichtert wird, und bewirkt tetanische Krämpfe,
die lebensbedrohlich sein können. Gegenmaßnahme: Erhöhung des Calciumspiegels (Calciumcarbonat i.v.). Diuretika können den Calciumhaushalt beeinflussen: Schleifendiuretika hemmen den Na/K/Cl-Transport, reduzieren die Aufladung in der Henle-Schleife und verringern die parazelluläre Ca++-Resorption Amilorid hemmt die apikale Natriumpermease (ENaC) der distalen Tubuli und fördert - so wie Thiazide - die Ca++-Resorption Thiazide hemmen die NaCl-Resorption, senken die zelluläre Na+-Konzentration, erhöhen das basolaterale Membranpotential und steigern damit die Ca++-Resorption
Diuretika können den Calciumhaushalt beeinflussen: Schleifendiuretika hemmen den Na/K/Cl-Transport, reduzieren die Aufladung in der Henle-Schleife und verringern die parazelluläre Ca++-Resorption Amilorid hemmt die apikale Natriumpermease (ENaC) der distalen Tubuli und fördert - so wie Thiazide - die Ca++-Resorption Thiazide hemmen die NaCl-Resorption, senken die zelluläre Na+-Konzentration, erhöhen das basolaterale Membranpotential und steigern damit die Ca++-Resorption
 Der Knochen speichert für seine Druckbelastbarkeit Calcium (~1 kg) und
Phosphat (~0,6 kg). Täglich werden ~500 mg Ca++
aus dem Knochen mobilisiert und wieder eingebaut (~0,05% des
Körperbestandes). Das Gleichgewicht zwischen Ab- und Einbau hängt von Faktoren ab wie physische Belastung (fördert
Mineralisierung), Ernährung, Alter, Geschlecht, hormonelle Regulation
Calcium und Phosphat werden reguliert durch Parathormon, (Thyreo-)
Calcitonin und Calcitriol (Vitamin D3-Hormon). Die Blutspiegel werden
vor allem durch
Parathormon und Calcitriol erhöht / aufrechterhalten (calciotrope
Hormone). Die Parathormonsekretion steigt bei sinkendem, und sinkt bei
steigendem Ca++-Spiegel. Der Ca++-Spiegel beträgt ~2,5 mMol/l (10
mg/dl) - knapp 60% freie Ionen (biologisch
wirksam und filtrierbar), gut 40% gebunden (Protein, Komplexsalze). Abnahme des freien Anteils erhöht die Erregbarkeit von
Muskelfasern und kann zu Tetanie führen Die Zellmembran der Hauptzellen der Epithelkörperchen hat
G-Protein-betriebene calciumsensitive Rezeptoren (CaSR), deren
Aktivierung die Synthese und Freisetzung von Parathormon hemmt. Parathormon steigert die Ca++-Resorption
in
Knochen und Nieren (distaler Tubulus), und fördert die Synthese von
Calcitriol, damit wirkt es der
Entmineralisierung des Knochens entgegen. Parathormon steigert die
renale Ausscheidung von Phosphat (geringere Resorption im proximalen
Tubulus). Die gesteigerte Phosphatausscheidung wirkt der Bildung von
Calciumphosphatkristallen außerhalb des Knochens entgegen. Primärer
Hyperparathyreoidismus kann zur Bildung von Nierensteinen führen
Der Parathormon-Effekt im Knochen hängt von der Einwirkungsdauer ab:
Kurze Parathormon-"Pulse" wirken anregend auf Osteoblasten,
längerdauernd wird die Resorption angeregt: RANKL nimmt zu,
Osteoprotegerin wird supprimiert, Osteoklasten werden angeregt
(erhöhtes turnover): Osteoblasten bilden osteoklastenanregende Zytokine, Grundsubstanz wird abgebaut, Calcium und Phosphat mobilisiert Calcitonin aus parafollikulären C-Zellen der Schilddrüse wird proportional zum Ca++-Spiegel
freigesetzt - gesteuert durch calciumsensitive Rezeptoren (CaSR). Calcitonin bremst Osteoklasten und regt Osteoblasten an
("Knochenschutzhormon"). Calcitonin verringert bei niedrigem Ca++-Spiegel
die Rückresorption von Phosphat im proximalen Nierentubulus sowie die
von Calcium im distalen Tubulus (synergistisch mit Parathormon). Calcitonin reagiert auf akute
Änderungen des Ca++-Spiegels
UV-B-Bestrahlung der Haut macht aus Dehydrocholesterin Prävitamin D;
dessen Hydroxylierung in Leber (25-Hydroxylase) und Niere
(1α-Hydroxylase) ergibt Calcitriol. Calcitriol (Vitamin D3) regt die
Resorption von Ca++ und HPO4--
aus dem Darm an. Parathormon und niedrige Ca/P-Spiegel steigern die Aktivität der
1α-Hydroxylase, hohe Ca/P-Spiegel senken sie. Die Leber bildet Vitamin
D-bindendes Protein (DBP), das wichtigste D3-Trägerprotein; ~0,4% des (biologisch aktiven) D3 ist
nicht proteingebunden (biologische
Halbwertszeit mehrere Stunden) D3 wirkt - vor allem in Dünndarm, Nierentubuli, Knochen und
Nebenschilddrüsen - über nukleäre Rezeptoren (VDR, Calcitriolrezeptor), der
an vitamin D-response elements
bindet und die Expression von Zielgenen steuert. D3 steigert die
Ca/P-Resorption im Dünndarm, fördert die Knochenmineralisierung und hat
zahlreiche weitere Wirkungen im Körper, z.B. stärkt es das Immunsystem Der Fibroblasten-Wachstumsfaktor 23 (FGF23) aus dem Knochen
steigert die Calciumresorption und Phosphatausscheidung in der Niere
Der Knochen speichert für seine Druckbelastbarkeit Calcium (~1 kg) und
Phosphat (~0,6 kg). Täglich werden ~500 mg Ca++
aus dem Knochen mobilisiert und wieder eingebaut (~0,05% des
Körperbestandes). Das Gleichgewicht zwischen Ab- und Einbau hängt von Faktoren ab wie physische Belastung (fördert
Mineralisierung), Ernährung, Alter, Geschlecht, hormonelle Regulation
Calcium und Phosphat werden reguliert durch Parathormon, (Thyreo-)
Calcitonin und Calcitriol (Vitamin D3-Hormon). Die Blutspiegel werden
vor allem durch
Parathormon und Calcitriol erhöht / aufrechterhalten (calciotrope
Hormone). Die Parathormonsekretion steigt bei sinkendem, und sinkt bei
steigendem Ca++-Spiegel. Der Ca++-Spiegel beträgt ~2,5 mMol/l (10
mg/dl) - knapp 60% freie Ionen (biologisch
wirksam und filtrierbar), gut 40% gebunden (Protein, Komplexsalze). Abnahme des freien Anteils erhöht die Erregbarkeit von
Muskelfasern und kann zu Tetanie führen Die Zellmembran der Hauptzellen der Epithelkörperchen hat
G-Protein-betriebene calciumsensitive Rezeptoren (CaSR), deren
Aktivierung die Synthese und Freisetzung von Parathormon hemmt. Parathormon steigert die Ca++-Resorption
in
Knochen und Nieren (distaler Tubulus), und fördert die Synthese von
Calcitriol, damit wirkt es der
Entmineralisierung des Knochens entgegen. Parathormon steigert die
renale Ausscheidung von Phosphat (geringere Resorption im proximalen
Tubulus). Die gesteigerte Phosphatausscheidung wirkt der Bildung von
Calciumphosphatkristallen außerhalb des Knochens entgegen. Primärer
Hyperparathyreoidismus kann zur Bildung von Nierensteinen führen
Der Parathormon-Effekt im Knochen hängt von der Einwirkungsdauer ab:
Kurze Parathormon-"Pulse" wirken anregend auf Osteoblasten,
längerdauernd wird die Resorption angeregt: RANKL nimmt zu,
Osteoprotegerin wird supprimiert, Osteoklasten werden angeregt
(erhöhtes turnover): Osteoblasten bilden osteoklastenanregende Zytokine, Grundsubstanz wird abgebaut, Calcium und Phosphat mobilisiert Calcitonin aus parafollikulären C-Zellen der Schilddrüse wird proportional zum Ca++-Spiegel
freigesetzt - gesteuert durch calciumsensitive Rezeptoren (CaSR). Calcitonin bremst Osteoklasten und regt Osteoblasten an
("Knochenschutzhormon"). Calcitonin verringert bei niedrigem Ca++-Spiegel
die Rückresorption von Phosphat im proximalen Nierentubulus sowie die
von Calcium im distalen Tubulus (synergistisch mit Parathormon). Calcitonin reagiert auf akute
Änderungen des Ca++-Spiegels
UV-B-Bestrahlung der Haut macht aus Dehydrocholesterin Prävitamin D;
dessen Hydroxylierung in Leber (25-Hydroxylase) und Niere
(1α-Hydroxylase) ergibt Calcitriol. Calcitriol (Vitamin D3) regt die
Resorption von Ca++ und HPO4--
aus dem Darm an. Parathormon und niedrige Ca/P-Spiegel steigern die Aktivität der
1α-Hydroxylase, hohe Ca/P-Spiegel senken sie. Die Leber bildet Vitamin
D-bindendes Protein (DBP), das wichtigste D3-Trägerprotein; ~0,4% des (biologisch aktiven) D3 ist
nicht proteingebunden (biologische
Halbwertszeit mehrere Stunden) D3 wirkt - vor allem in Dünndarm, Nierentubuli, Knochen und
Nebenschilddrüsen - über nukleäre Rezeptoren (VDR, Calcitriolrezeptor), der
an vitamin D-response elements
bindet und die Expression von Zielgenen steuert. D3 steigert die
Ca/P-Resorption im Dünndarm, fördert die Knochenmineralisierung und hat
zahlreiche weitere Wirkungen im Körper, z.B. stärkt es das Immunsystem Der Fibroblasten-Wachstumsfaktor 23 (FGF23) aus dem Knochen
steigert die Calciumresorption und Phosphatausscheidung in der Niere |
